
Abstract
A rice PAL (phenylalanine ammonia-lyase) gene sequence (rPAL-P5), which is highly similar to and likely the same as a previously described rice ZB8PAL gene, including the 5′-upstream and exon I coding regions of PAL, was isolated using PCR amplification. The expression of several PALs, including rPAL-P5, was strongly induced following inoculation with Pyricularia oryzae or treatment with a P. oryzae elicitor. To identify the promoter region induced by the P. oryzae elicitor, we constructed and subsequently transformed rPAL-P5 promoter deletion series into rice calli using particle bombardment. Results from both elicitor-inducible reporter gene and gel mobility shift assays demonstrated that the sequence −349 to −256 of the rPAL-P5 promoter includes a cis-element involved in the induction of P. oryzae.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Secondary metabolites such as phytoalexins, lignins and flavonoids produced by the phenylpropanoid pathway may help protect higher plants from pathogen attack (Dixon and Pavia 1995). Phenylalanine ammonia-lyase (PAL) is a key enzyme in this pathway, catalyzing the first committed step in phenylpropanoid biosynthesis, namely, the deamination of phenylalanine to yield trans-cinnamic acid and NH4 + (Bate et al. 1994; Howles et al. 1996).
PAL genes are induced by pathogen infection and specific elicitor molecules (Hughes et al. 1989; Sharan et al. 1998). The promoter sequences responsible for this induction have been identified in some genes, including parsley PAL-1, pea PSPAL-1, carrot gDcPAL1 and others (Silva et al. 1993; Yamada et al. 1994; Takeda et al. 2002). Many of these cis-elements share a conserved AC-rich element, the mutation or deletion of which can result in a reduction or loss of induced activation following pathogen attack (Lois et al. 1989; Seki et al. 1997, 1999; Murakami et al. 1997).
Relatively few studies have dealt with monocot PAL genes. To date, only PALs in rice (Minami et al. 1989; Minami and Tanaka 1993; Zhu et al. 1995), wheat (Liao et al. 1996) and barley (Kervinen et al. 1998) have been cloned, and only rice ZB8PAL and barley hpal2, -3,-4, -6 have been shown to be induced following pathogen infection. No further analysis of these promoters has been reported.
We report here the induction of the rice rPAL-P5 gene following Pyricularia oryzae infection or elicitor treatment and identify a region of the promoter involved in this induction.
Materials and methods
Plant material, pathogen and vectors
Seeds of rice (Oryza sativa L. cv. Aizhixu) and fungal strain of Pyricularia oryzae cv. 131 were kindly provided by Dr. Peng Youliang (China Agricultural University). Seedlings of O. sativa L. cv. Aizhixu were grown in a heated greenhouse (28–32°C). The induction and culture of rice calli were performed as previously described by Yuan et al. (2002). T-vector pUCm-T was purchased from the Shanghai Sangon Company. The plant transformation vectors pCambia1381Xa (hygromycin R, GUS-NOS) and pCambia1301 (hygromycin R, 35S-GUS-NOS) were kindly provided by Dr. Richard Jefferson.
Primers and PCR for rPAL-P5 and 5′-truncated promoters
Based on the nucleotide sequence of ZB8PAL, the rPAL-P5 DNA fragment (position −956 to +466) containing the putative promoter and a part of PAL exon I was generated using primers rPP-a (5′) (−956: 5′-TCGCGCCTACTGACCTACGTAT-3′) and PP-1 (3′) (+447 to +466: 5′-GGTGGTGACACCGTAGCTGT-3′) (Fig. 1). The 5′ truncated promoter segments from rPAL-P5 were obtained using rPP-a, rPP-b (−646: 5′-CATTCTGAATAGGGCATGCA-3′), rPP-c (−424: 5′-CACACCAACTTCCATCCATA-3′), rPP-d (−264: 5′-AGACGCAGCAAGTCCGTCGC-3′), rPP-e (−45: 5′-ACCCACCCGCCTATTTAAGC-3′) and PP-a (3′) (+126 to +145: 5′-AAGCTTGCACTCCATTTCAGTACCAG-3′ with a terminal HindIII site) (Fig. 3). The transcriptional start site was at +1, and the “A” residue of the translational start site ATG was at +137.

Comparison of rPAl-P5, ZB8PAL and contig6019 nucleotide sequences. The sequence of rPAL-P5 is shown on the top line. Bases in ZB8PAL and contig6019 common to all three nucleotides are denoted with dots, with only the variable bases indicated for the former two sequences. Contig6019 is from the rice genome sequences of HuaDa, China. The transcription start site “A” in the shaded area is at the +1 position. The codon representing translational initiation is indicated by a rectangle, and the “A” of ATG is at the +137 position. Arrow Primers (rPP-a and PP-1) for rPAL-P5
Rice genomic DNA was extracted from 1 g of leaves of 1-month old plants by the CTAB method (Doyle et al. 1990). PCR parameters were 35 cycles of pre-denaturing at 94°C for 120 s, denaturing at 94°C for 60 s, annealing at 60°C for 70 s, enzymatic reaction at 72°C for 90 s. The samples were then kept at 72°C for 10 min.
Elicitation of rice calli
After P. oryzae had been cultured on solid oat-dextrose medium at 28°C in the dark for 7–8 days, the mycelium was washed with 4 ml water from the solid medium, transferred to 200 ml oat liquid medium and incubated at 28°C in the dark for 4–5 days. The mycelium was then harvested by centrifugation at 10,000 g for 5 min, washed several times with water, homogenized in 20 ml water using a glass homogenizer and then centrifuged at 10,000 g for 5 min. The supernatant was autoclaved and stored at −70°C.
For the elicitation of rice calli, approximately 1 g of rice calli was treated at 25°C for 1, 3, 6 and 12 h in the dark with a five-fold diluted crude elicitor solution, or with water as a control. The treated rice calli were frozen in liquid nitrogen at each time point and stored at −70°C until used for RNA extraction.
RNA isolation and Northern blot hybridization
At each time point, total RNA was isolated from rice calli treated with crude elicitor using the RNeasy Plant Mini kit (QIAGEN, Valencia, Calif.) and then electrophoresed through a 1.2% denaturing agarose gel (10 μg per lane). A 136-bp specific untranslated sequence of rPAL-P5 was used as a probe, and Northern blot hybridization was carried out according to the Bio-Rad Zeta-GT membrane manual (Bio-Rad, Hercules, Calif.).
Construction of chimeric genes, transformation and selection of rice calli
The rPAL-P5 promoter (−956 to +145) and its several 5′ truncated promoter segments obtained by PCR were cloned into the pUCm-T vector and sequenced by the Sangon Company. These fragments were then excised and ligated to GUS in pCambia1381Xa (Fig. 3). All open reading frames (ORFs) of rPP-GUS chimeric genes were identified by DNA sequencing. The series of rPP-GUS chimeric genes were then transformed into rice calli by particle bombardment, selected on N6 medium containing 50 mg/l hygromycin (Zheng et al. 1997; Yuan et al. 2002) and then PCR-amplified (data not shown).
Fluorometric GUS assay of elicitor-treated transgenic rice calli
Transgenic calli were cut into small pieces and cultured in N6 liquid medium containing 2 mg/l 2,4-D and 50 mg/l hygromycin. The calli were treated for 8 h with a five-fold diluted crude elicitor solution or water with gentle agitation at 100 rpm, at 25°C in the dark. The GUS activity of rice calli, determined using 4-MUG according to the procedure of Jefferson (1987), is presented as 4-MU pmol/mg per protein per minute.
Preparation of nuclear proteins
Rice calli treated with elicitor or water for 0, 1, 1.5 and 2 h were harvested individually. Nuclear proteins were extracted as described by Nagao (1993) with some modifications: (1) the concentration of protease inhibitors in all buffers was doubled; (2) the supernatant was separated from the pellet produced by 45% saturation with ammonium sulfate, then continued to be brought to 85% saturation. These precipitated proteins were collected by centrifugation at 14,000 rpm for 10 min. The Bradford Bio-Rad Protein Assay kit was used for the determination of nuclear protein. Aliquots of extract were stored at −70°C until used.
Gel mobility shift assays
Two DNA fragments, RP-a (−424 to −326 ) and RP-b (−349 to −256), were amplified by PCR and cloned into the pBS vector to construct the recombination plasmids pQRP-a and pQRP-b, respectively. The primers used for RP-a were rPPc (5′-CACACCAACTTCCATCCATA-3′) and PP2D-3 (5′-GGGAAGCTTCTACTTTTGGTACTCCTC-3′, containing a terminal HindIII site), while the primers used for RP-b were PP2D-5 (5′-GGGAAGCTTGACGAGGAGTACCAAAAGT-3′, containing a terminal HindIII site) and dp-1r (5′-TTGCTGCGTCTGCGGCTGCC-3′). RP-a and RP-b were then excised from pQRP-a and pQRP-b by HindIII digestion, and 60 ng DNA of each was radiolabeled at the terminus using α-[32P]-dCTP and Klenow DNA polymerase. The labeled DNA fragments were precipitated with NH4OAc and ethanol at −70°C for 1.5 h.
Gel mobility shift assays were performed using the protocol described by Nagao et al. (1993). Unlabeled RP-b fragments and unlabeled unrelated DNA fragments were used as DNA competitors or non-competitors, respectively. The protein/DNA complexes were analyzed by electrophoresis through a 5% native polyacrylamide gel (29:1 cross-linking ratio) in 1× TBE buffer. Following electrophoresis, the gel was dried and autoradiographed.
Results
Cloning of rPAL-P5 gene
A specific DNA fragment was amplified from rice genomic DNA with the PCR primers rPP-a and PP-1. Sequencing showed that this fragment contained a gene (named rPAL-P5) which was 99.2% similar to the rice ZB8PAL sequence (Zhu et al. 1995) and included 956 bp of promoter sequence, a 136-bp 5′ untranslated region and 330 bp of exon I. Furthermore, rPAL-P5 displayed significant sequence homology (99.7%) to the DNA sequence contig6019 of the rice genome from 5,987 to 5,032 (Fig. 1). The fact that rPAL-P5 and ZB8PAL shared significant sequence homology and that they were both located at the same site (Contig6019) in the rice genome indicated that rPAL-P5 and ZB8PAL were the same gene but that each contained a few different bases, probably as a result of varied rice breeds. Furthermore, blasting in the rice genome indicated that the promoter and 5′ untranslated region of rPAL-P5 were specific and present only as a single copy in the rice genomic DNA.
Induction of rPAL-P5 gene by the elicitor
Consistent with other reports, the induction of PAL genes was detected in rice plants infected with P. oryzae or in rice calli treated with elicitor. Following transcription, PAL enzyme activity increased markedly and reached a maximum 8 h following the initiation of elicitor treatment in rice calli (data not shown). In an effort to analyze the specific induction of rPAL-P5 by elicitor, we treated rice calli with elicitor and subsequently carried out Northern blot RNA analysis using the 136-bp rPAL-P5 untranslated sequence as the probe (Fig. 2). rPAL-P5 transcripts were present 1 h after induction and accumulated to a maximum level—about 20-fold that of the control—3 h post-induction. By 6 h, rPAL-P5 mRNA levels were lower than 50% that observed at 3 h, and by 12 h, they were nearly identical to that of the controls.

Induction of rPAL-P5 by the Pyricularia oryzae elicitor. rPAL-P5, a specific DNA fragment representing 136 bp of an untranslated sequence in rPAL-P5, was used as a probe; 18S, part of a cDNA derived from 18S RNA was used as a probe
Elicitor induction of rPP-GUS chimeric genes in rice calli
In an effort to analyze the presence of a functional cis-acting region associated with the induction by pathogen elicitor, we constructed chimeric genes containing the rPAL-P5 promoter or 5′ promoter deletions of rPAL-P5, as shown in Fig. 3, and introduced these into rice calli using particle bombardment. PCR confirmed the presence of chimeric rPP-GUS genes in the transformed calli (data not shown).

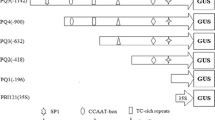
Effects of the P. oryzae elicitor on the expression of rPP-GUS chimeric genes. A schematic representation of the various chimeric constructs used is shown on the left. All of these were inserted into pCambia1381Xa 5′ to GUS-NOS. The E/W ratios reflecting GUS enzyme activity due to different chimeric genes in the transgenic rice calli are shown on the right. E Treatment with elicitor for 8 h, W treatment with water for 8 h. Ratios were from at least three independent transgenic clones. Error bars Standard deviation. GUS activity was detected using a fluorometric assay
The basal expression levels of GUS activity differed greatly amongst the different clones, which may be due to variable copy numbers being introduced into the latter using the particle bombardment method. The GUS activity ratios between elicitor and water treatments (E/W ratio) for the same rPP-GUS chimeric gene were similar. Consequently, the effects of elicitor on the expression of different rPP-GUS chimeric genes were arrived at by calculating the E/W ratios of GUS activity, as shown Fig. 3. Since PAL activity in rice calli reached a maximum 8 h following elicitor treatment (data not shown), we elected to examine the GUS activity of transgenic calli at this time point. A construct lacking a promoter (GUS-NOS) was used as a negative control, and the 35S-GUS-NOS construct was used as positive control. The negative control showed no GUS activity, while the E/W ratio for the positive control construct was approximately 1. The E/W ratios of transgenic calli with rPPa-GUS, rPPb-GUS or rPPc-GUS were about 3, indicating that the elicitor induced gene expression threefold. These results suggest that the −956 to −424 region is not involved in induction. While the E/W ratios of rPPd-GUS and rPPe-GUS were only about 1.6 and 1.1, respectively, which suggests that the absence of the −424 to −264 or −264 to −45 regions could cause a reduction in the induction of the rPAL-P5 promoter and that the −424 to −264 region plays a greater role in decreasing the induction than the −264 to −45 region, it is suggested that the −424 to −264 region is more important for the elicitor-induced response of rPAL-P5.
Elicitor-induced binding assay of the cis-region of the rPAL-P5 promoter
Although two regions (−424 to −264 and −264 to –45) were associated with the induction of the rPAL-P5 promoter, the −424 to –264 region seemed to have a greater effect. In an effort to further characterize this region, we broke down these regions into two fragments—RP-a (−424 to −326) and RP-b (−349 to −256)—and analyzed protein binding using a gel mobility shift assay. As shown in Fig. 4A, complexes were only formed when the RP-b fragment was mixed with elicitor-treated nuclear extracts, with maximum binding appearing 2 h following elicitor treatment (Fig. 4B). In addition, the complexes were enhanced by increasing the levels elicitor-treated nuclear extracts and inhibited by a 25- or 50-fold excess of competitor (unlabeled RP-b fragments) but not by non-competitors (unlabeled unrelated DNA fragments) (Fig. 4C). These findings reflect the presence of binding sites for elicitor-induced nuclear factors within the –349 to −256 region, which is a cis-region involved in the activation of the rPAL-P5 promoter by the P. oryzae elicitor.

Binding of nuclear extracts to DNA fragments of the rPAL-P5 promoter. E Nuclear extracts from elicitor-treated rice calli, W nuclear extracts from water-treated rice calli, RP-a −424 to –326 fragment, RP-b −349 to –256 fragment, C specific competitors, N nonspecific competitors. The amount of labeled probe used was 1 ng for each lane. A The amount of nuclear extract used from rice calli treated with E or W for 2 h was 2 μg for each lane, B the amount of nuclear extract used for the different time points was 2 μg for each lane, C competitors were unlabeled RP-b fragments
Discussion
Analysis of 5′ deleted promoters suggested that the −424 to -265 region is important for the elicitor induction of rPAL-P5 (Fig. 3). Further, gel mobility shift assays showed that the −349 to -256 region of the rPAL-P5 promoter could form specific DNA-protein complexes with nuclear extracts from rice calli treated with P. oryzae crude elicitor, indicating that the −349 to −256 region might contain cis-elements responsible for elicitor induction (Fig.4). AC-rich elements have been reported to be important for pathogen or elicitor induction of PALs and other genes of the phenylpropanoid pathway (such as CHS and 4CL) (Douglas et al. 1987; Seki et al. 1999) in several dicotyledonous plants, but this has not yet been reported for monocotyledonous plants. We failed to find putative AC-rich elements in the cis-region (−349 to –256) of the rPAL-P5 promoter, suggesting that cis-elements different from the AC-rich sequence might be present in this region.
The identification of PAL regulatory proteins associated with pathogen activation is very important for furthering our understanding of the regulatory mechanisms that PALs play in terms of plant defense responses. To date, only a few plant regulatory proteins have been reported. In parsley, the BPF-1 (BoxP-binding Factor) protein, which binds specifically to BoxP of the PcPAL promoter, has been cloned, and it was found that the BPF-1 gene is activated by elicitor and co-expressed with PAL at infection sites (Silva et al. 1993). Sugimoto et al. (2000) found that tobacco NtMYB2 can interact with BoxL of the PAL promoter, that the gene encoding NtMYB2 is activated by pathogen and elicitor and that the constitutive expression of NtMYB2 stimulates the expression of PAL in uninfected tobacco plants. In addition, the MAPK (mitogen-activated protein kinase) cascade reaction (NtMEK2-SIPK/WIPK) was reported to be associated with the expression of PALs and probably takes part in the regulation of defense-related genes, including PALs, in plants (Yang et al. 2001). The regulation of PALs in plants appears to be quite complicated and much remains unknown, especially in the case of monocotyledonous plants. We are presently trying to isolate and identify the regulatory factor of rPAL-P5 that is activated following pathogen infection in rice plant.
Abbreviations
- CTAB :
-
Cetyltrimethylammonium bromide
- 2,4-D :
-
2,4-Dichlorophenoxyacetic acid
- GUS :
-
β-Glucuronidase
- 4-MU :
-
4-Methylumbelliferone
- 4-MUG :
-
4-Methylumbelliferyl glucuronide
- NOS :
-
Nopaline synthase
- PAL :
-
Phenylalanine ammonia-lyase
References
Bate NJ, Weiting JO, Meromi A, Nadler-Hassar T, Doerner PW, Dixon RA, Lamb CJ, Elkind Y (1994) Quantitative relationship between phenylalanine ammonia-lyase levels and phenylpropanoid accumulation in transgenic tobacco identifies a rate-determining step in natural product synthesis. Proc Natl Acad Sci USA 9108:7608–7612
Dixon RA, Paiva NL (1995) Stress-induced phenypropanoid metabolism. Plant Cell 7:1085–1097
Douglas C, Hoffmann H, Schulz W, Hahlbrock K (1987) Structure and elicitor or UV-light-stimulated expression of two 4-coumarate:CoA ligase genes in parsley. EMBO J 6:1189–1195
Doyle JJ, Doyle JL, Hoztorium BLH (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Howles PA, Sewalt VJH, Paiva NL, Elkind Y, Bate NJ, Lamb C, Dixon RA (1996) Over-expression of l-phenylalanine ammonia-lyase in transgenic tobacco plants reveals control points for flux into phenylpropanoid biosynthesis. Plant Physiol 112:1617–1624
Hughes RK, Dickerson AG (1989) The effect of ethylene on phenylalanine-lyase (PAL) induction by a fungal elicitor in Phaseolus vulgaris. Physiol Mol Plant 34:361–378
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusion: β-glucuronidase as a sensitive and versatile gene-fusing marker in higher plants. EMBO J 6:3901–3907
Kervinen BT, Peltonen S, Teeri TH, Karjalainen R (1998) Differential expression of phenylalanine ammonia-lyase genes in barley induced by fungal infection or elicitors. New Phytol 139:293–300
Liao YC, Li HP, Kreuzaler F, Fischer R (1996) Nucleotide sequence of one of two tandem genes encoding phenylalanine ammonia-lyase in Triticum aestivum (PGR96-102). Plant Physiol 112:1398–1398
Lois R, Dietrich A, Hahlbrock K, Shulz W (1989) A phenylalanine ammonia-lyase gene from parsley: structure, regulation and identification of elicitor- and light-responsive cis-acting elements. EMBO J 8:1641–1648
Minami E, Tanaka Y (1993) Nucleotide sequence of the gene for phenylalanine ammonia-lyase of rice and its deduced amino acid sequence. Biochim Biophys Acta 1171:321–322
Minami E, Ozaki Y, Matsuoka M, Koizuka N, Tanaka Y (1989) Structure and some characterization of the gene for phenylalanine ammonia-lyase from rice plants. Eur J Biochem 185:19–25
Murakami Y, Ichinose Y, Shiraishi T, Yamada T (1997) Functional analysis of the putative cis-elements Involved in activation by elicitor or UV and deactivation by suppressor in the promoter of a pea gene for phenylalanine ammonia-lyase. Plant Cell Physiol 38:1403–1408
Nagao RT, Goekjian VH, Hong JC, Key JL (1993) Identification of protein-binding DNA sequences in an auxin-regulated gene of soybean. Plant Mol Biol 21:1147–1162
Seki H, Ichinose Y, Ito M, Shiraishi T, Yamada T (1997) Combined effects of multiple cis-acting elements in elicitor-mediated activation of PSCHS1 gene. Plant Cell Physiol 38:96–100
Seki H, Nagasugi Y, Ichinose Y, Shiraishi T, Yamada T (1999) Changes in in vivo DNA-protein interactions in pea phenylalanine ammonia-lyase and chalcone synthase gene promoter induced by fungal signal molecules. Plant Cell Physiol 40:88–95
Sharan M, Taguchi G, Gonda K, Jouke T, Shimosaka M, Hayashida N, Okazaki M (1998) Effects of methyl jasmonate and elicitor on the activation of phenylalanine ammonia-lyase and the accumulation of scopoletin and scopolin in tobacco cell cultures. Plant Sci 132:13–19
Silva OC, Klein L, Schmelzer E, Trezzini GF, Hahibrock K (1993) BPF-1, a pathogen-induced DNA-binding protein involved in the plant defense response. Plant J 4:125–135
Sugimoto K, Takeda S, Hirochika H (2000) MYB-related transcription factor NtMYB2 induced by wounding and elicitors is a regulator of the tobacco retrotransposon Tto1 and defense-related genes. Plant Cell 12:2511–2528
Takeda J, Ito Y, Maeda K, Ozeki Y (2002) Assignment of UVB-responsive cis-element and protoplastization (dilution-) and elicitor-responsive ones in the promoter region of a carrot phenylalanine ammonia-lyase gene (gDcPAL1). Photochem Photobiol 76:232–238
Yamada T, Sriprasertsak P, Kato H, Hashimoto T, Shimizu H, Shiraishi T (1994) Functional analysis of the promoters of phenylalanine ammonia-lyase genes in pea. Plant Cell Physiol 35:917–926
Yang KY, Liu Y, Zhang S (2001) Activation of a mitogen-activated protein kinase pathway is involved in disease resistance in tobacco. Proc Natl Acad Sci USA 98:741–746
Yuan HY, Ming XT, Wang LJ, Hu P, An CC, Chen ZL (2002) Expression of a gene encoding trichosanthin in transgenic rice plants enhances resistance to fungus blast disease. Plant Cell Rep 20:992–998
Zheng HH, Li Y, Yu ZH, Li W, Chen MY, Ming XT, Casper R, Chen ZL (1997) Recovery of transgenic rice plant expressing the rice dwarf virus outer coat protein gene (S8). Theor Appl Genet 94:522–527
Zhu Q, Dabi T, Beeche A, Yamamoto R, Lawton MA, Lamb C (1995) Cloning and properties of a rice gene encoding phenylalanine ammonia-lyase. Plant Mol Biol 29:535–550
Acknowledgements
We thank Prof. Youliang Peng of China Agricultural University for providing the rice seeds (Oryza sativa L. cv. Aizhixu) and the fungal strain of Pyricularia oryzae cv. 131. This work was supported by a grant from the National Natural Science Foundation (grant no. 39980003) and the National Key Basic Research “973” Program (grant no. G2000016204) of China.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J.C. Register III
Rights and permissions
About this article
Cite this article
Wang, L., An, C., Qian, W. et al. Detection of the putative cis-region involved in the induction by a Pyricularia oryzae elicitor of the promoter of a gene encoding phenylalanine ammonia-lyase in rice. Plant Cell Rep 22, 513–518 (2004). https://doi.org/10.1007/s00299-003-0717-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-003-0717-3