
Abstract
In this transglobal, randomized, double-blind, placebo-controlled, treat-to-target study, the maintenance of efficacy was compared between biologic–and biologic-free–disease-modifying antirheumatic drug (DMARD) combination regimens after low disease activity (LDA) was achieved with biologic DMARD induction therapy. Patients with moderate-to-severe rheumatoid arthritis despite methotrexate therapy received open-label etanercept 50 mg subcutaneously once weekly plus methotrexate with or without other conventional synthetic (cs) DMARDs for 24 weeks. Patients achieving LDA [disease activity score in 28 joints based on erythrocyte sedimentation rate (DAS28-ESR) <3.2] at week 24 were randomized to receive etanercept–methotrexate combination therapy or placebo–methotrexate combination therapy, with or without other csDMARDs, for 28 weeks. In the open-label period, 72% of patients achieved DAS28-ESR LDA at week 24. Patients enrolled in the double-blind period had long-standing rheumatoid arthritis and high disease activity at baseline (mean duration, 8.1 years; DAS28-ESR, 6.4). In the etanercept and placebo combination groups, 44% versus 17% achieved DAS28-ESR LDA and 34 versus 13% achieved DAS28-ESR remission at week 52 (p < 0.001). Adverse events were reported in 37 and 43%, serious adverse events in 0 and 4%, and serious infections in 0 and 2% in these groups, respectively, in the double-blind period. After induction of response with etanercept combination therapy following a treat-to-target approach in patients with long-standing rheumatoid arthritis and high disease activity at baseline, the etanercept combination regimen was significantly more effective in maintaining LDA and remission than a biologic-free regimen.
ClinicalTrials.gov identifier. NCT01578850.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In individuals with rheumatoid arthritis (RA), joint destruction often starts within months of symptom onset and is evident radiographically within 2 years [1, 2]. Because control of inflammation may reduce joint damage and functional disability, new treatment guidelines recommend early treatment of patients with RA to induce low disease activity (LDA) or clinical remission, followed by less intensive treatment to maintain this response [3]. Treatment with anti-tumor necrosis factor agents and the conventional synthetic disease-modifying antirheumatic drug (csDMARD) methotrexate has been shown to significantly improve clinical, radiographic, and functional outcomes in patients with early RA [4,5,6,7]. However, treatment guidelines and clinical trials with rigid patient inclusion criteria and treatment protocols may have limited application in real-world practice. In addition, relatively few studies have been conducted to assess treat-to-target (T2T) strategies in RA patients from geographic regions outside the United States and Western Europe, where biologic therapy is frequently limited to use in severe and refractory disease.
To address these gaps in knowledge, a transglobal T2T study was conducted that compared the maintenance of efficacy provided by etanercept and methotrexate with or without other csDMARDs versus placebo and methotrexate with or without other csDMARDs in patients with moderate-to-severe RA who had achieved LDA with biologic combination induction therapy (ClinicalTrials.gov identifier: NCT01578850).
Materials and methods
Study design
This report presents findings from prespecified analyses for this two-period, randomized, double-blind, placebo-controlled study that was conducted from July 2012 to March 2015 at 61 centers in 19 countries in Africa, Asia, Central and Eastern Europe, Latin America, and the Middle East. The study was conducted according to the International Conference on Harmonisation guidelines for Good Clinical Practice (GCP), the International Ethical Guidelines for Biomedical Research Involving Human Subjects from the Council for International Organizations of Medical Sciences, and the Declaration of Helsinki. The study protocol and informed consent form were approved by the independent ethics committee or institutional review board at each participating center before patient screening.
Patients
Adults were eligible for enrollment in the initial 24-week open-label induction period if they had active moderate-to-severe RA, defined as a disease activity score in 28 joints [DAS28; on the basis of erythrocyte sedimentation rate (ESR)] of ≥3.2, with ≥6 tender joint count and ≥6 swollen joint count or an ESR level ≥28 mm/h, and a C-reactive protein (CRP) level ≥3.5 mg/L despite methotrexate therapy. Patients had received a diagnosis of RA at least 1 year prior to screening, which was based on the 1987 revised classification criteria of the American College of Rheumatology (ACR) [8]. Participants were required to have been receiving methotrexate therapy administered in a dose of ≥10 mg/week for at least 12 weeks before enrollment and at stable doses for at least 4 weeks before screening. Nonsteroidal anti-inflammatory drug (NSAID) dose must have been stable for at least 2 weeks and the prednisone (or equivalent) dose ≤7.5 mg/day and stable for 4 weeks before the baseline visit.
Patients were excluded if they had received a csDMARD other than methotrexate, sulfasalazine, hydroxychloroquine, or leflunomide or a biologic DMARD within 2 months of baseline, or a biologic B cell-depleting agent within 2 years of baseline. Patients were also excluded if they had received any live vaccine within 4 weeks of baseline or during the study or had active tuberculosis in the previous 2 years. Patients with latent tuberculosis were included only when local guidelines for prophylactic treatment were followed and if such treatment was initiated or completed within 1 month of baseline. All patients provided written informed consent before initiation of all study-related procedures.
In the subsequent 28-week, double-blind, placebo-controlled maintenance period, participants were eligible for randomization if they had completed the open-label period, had achieved LDA (DAS28-ESR <3.2) at week 24, and were willing to continue stable doses of all concomitant treatments for RA received at week 24 through week 52 (unless dose adjustment was needed because of adverse events). Patients were excluded from this period if they had received a prednisone dose of >7.5 mg/day or the dose had been changed within 14 days of randomization (week 24), they had received a modified methotrexate dose within 8 weeks of randomization (with the exception of dose reductions because of adverse events), or they had taken any prohibited medications.
Study treatment
Patients who were enrolled in the initial open-label period received subcutaneous injections of 50 mg of etanercept weekly plus oral methotrexate with or without the other permitted csDMARDs sulfasalazine, hydroxychloroquine, and leflunomide for 24 weeks. Doses of the csDMARDs were adjusted up to week 16 at the investigator’s discretion with the goal of patients achieving remission (DAS28-ESR <2.6). After week 16, doses of these agents were to remain stable unless patients had unacceptable adverse events. Patients who did not achieve the criteria for LDA (DAS28-ESR <3.2) at week 24 of the open-label period were considered not to have had a response and were discontinued from the study.
Patients who met the criteria for LDA in the open-label period were randomly assigned to one of two treatment groups: a group that received etanercept at a dose of 50 mg administered as a weekly subcutaneous injection plus oral methotrexate with or without other csDMARDs (etanercept combination-therapy group) or a group that received a weekly placebo subcutaneous injection plus oral methotrexate with or without other csDMARDs (placebo combination-therapy group) for 28 weeks. In both groups, patients maintained stable doses of methotrexate, other csDMARDs, and corticosteroids throughout the second period, although reductions in doses were allowed at the investigator’s discretion in patients with adverse events.
In the double-blind period, patients in the placebo combination-therapy group who experienced a flare, defined as a loss of LDA (DAS28-ESR ≥3.2) and worsening in DAS28-ESR ≥0.6, received etanercept 50 mg weekly as rescue therapy in a blinded fashion. Patients in the etanercept combination-therapy group who met criteria for flare continued to receive their randomized treatment in a blinded fashion.
Blinding and randomization
In the second period, the study was patient-, investigator-, and sponsor-blinded. Pre-filled syringes were labeled, such that patients’ treatment assignment could not be determined. Blinding was only broken in emergency situations involving patient safety; treatment and randomization information was otherwise kept confidential and not released to investigators or study staff until the study’s conclusion. Methotrexate was used open label in both study periods, with bottles labeled to allow identification of contents.
Screening, enrollment, and randomization were accomplished using an automated internet/telephone randomization system (i.e., the Interactive Web Response System). At the screening visit, the investigative site contacted the system to screen the patient. At week 24, the system associated the patient with the next available treatment on the randomization schedule and provided a randomization number; patients who met LDA criteria were subsequently randomized in a 1:1 ratio to one of the two treatment groups.
Collected patient data and assessments
The primary efficacy endpoint was the percentage of patients in the etanercept and placebo combination-therapy groups in the double-blind period who achieved DAS28 LDA based on ESR (DAS28-ESR <3.2) at week 52 without rescue medication. Secondary endpoints included DAS28 remission based on ESR and C-reactive protein (DAS28-ESR/-CRP <2.6); DAS28 LDA based on CRP (<3.2); LDA and remission according to simplified disease activity index criteria (SDAI; LDA, SDAI >3.3 to ≤11; remission, SDAI ≤3.3) and clinical disease activity index criteria (CDAI; LDA, CDAI >2.8 to ≤10; remission, CDAI ≤2.8); and American College of Rheumatology (ACR) 20, ACR50, ACR70, and ACR90 responses (20, 50, 70, and 90% reduction, respectively, in the number of tender and swollen joints and improvement in three other ACR core set variables). Changes from baseline in DAS28, SDAI, CDAI, swollen and tender joint counts (0–28 joints), physician and patient global assessments (1–10 numerical rating scale), ESR and CRP levels, and patient-assessed general health and pain (100-mm visual analog scales) were assessed. Patient-reported outcomes were also evaluated with the Health Assessment Questionnaire-Disability Index (HAQ-DI; scores 0–3, with higher scores denoting greater disability), and with the EuroQoL-5 health state visual analog scale and utility total index.
In the double-blind period, the proportions of patients who experienced a flare were assessed, with flare defined as DAS28-ESR ≥3.2 and an increase of ≥0.6 from the week 24 DAS28-ESR score. Time to flare was also investigated in the second period. To assess if patients who achieved an early response to open-label treatment were more likely to also respond to double-blind treatment, the proportions of patients who achieved both DAS28 LDA at week 16 and the primary endpoint at week 52 were calculated in each of the randomized treatment groups.
Statistical analysis
Efficacy analyses were conducted in the full analysis set (FAS) population in each period using the last observation before rescue carried-forward (LOCF) approach. For the open-label period, the FAS population included patients who had taken at least one dose of study medication during the period; for the double-blind period, it included randomized patients who had achieved DAS28-ESR <3.2 at week 24, had received at least one dose of study medication, and had at least one DAS28-ESR evaluation in the period. Due to noncompliance with GCP at one study center, 11 patients were excluded from the efficacy analyses for the open-label period and 6 patients for the double-blind period. Analysis of covariance and a Cochran-Mantel-Haenszel test were used for between-group comparisons of continuous and binary data, respectively, at week 52. For all efficacy and patient-reported outcome analyses in the double-blind period in which LOCF imputation was applied, for patients receiving rescue therapy, the final value before the first dose of rescue therapy was used as the “carried-forward” value. Safety findings were analyzed in all patients who had received at least one dose of study medication during each period.
Time to flare during the double-blind period was analyzed using Kaplan-Meier and log-rank tests. For patients who experienced flare, the time to flare was measured from randomization until the occurrence of the first flare. All other patients were censored and time to event was measured from randomization until the last observed visit in the double-blind period.
In post hoc analyses of the effect of early treatment response in the open-label period, Chi-square tests were used to compare the subgroups of patients who did and did not achieve early LDA, both within each treatment group and overall stratified by treatment group. Treatment was also compared within each early response subgroup separately and overall stratified by early response subgroup. The Breslow-Day test was used to test for treatment by early response interaction.
Results
Patient disposition and baseline characteristics
Patient disposition through the course of the study is shown in Fig. 1. Of the 489 patients treated in the open-label period, 343 received treatment in the double-blind period (etanercept combination-therapy group, n = 167; placebo combination-therapy group, n = 176; safety population); 331 patients were included in efficacy analyses in the double-blind period (n = 163 and n = 168, respectively; FAS); and 316 patients completed the study (n = 154 and n = 162). Of patients treated in the double-blind period, the proportions of patients who discontinued treatment for safety and nonsafety reasons were similar between the treatment groups.

Patient disposition in the open-label and double-blind periods
Demographic and disease characteristics were comparable between the treatment groups at baseline of the double-blind period (Tables 1, 2). Patients who were analyzed in the double-blind period had long-standing RA (mean duration, 8.1 years) and high disease activity (DAS28-ESR, 6.4).
Efficacy
Response and other efficacy outcomes in the open-label period
Among patients enrolled in the open-label period, who all received etanercept combination induction therapy, 341 and 126 (72 and 27% of 473 patients included in this analysis) achieved LDA or remission, respectively, according to DAS28-ESR criteria at week 24. A total of 342 and 66 patients (73 and 14% of 472 patients analyzed) had LDA or remission based on SDAI criteria, and 358 and 51 (76 and 11% of 473 patients analyzed) had LDA or remission based on CDAI criteria. ACR20, ACR50, ACR70, and ACR90 responses were achieved, respectively, by 412, 340, 185, and 27 patients (88, 73, 39, and 6% of 469 patients analyzed) in this period. At week 24, 377 and 221 patients (80 and 47% of 473 patients analyzed) achieved a minimal clinically important difference in HAQ-DI (improvement >0.22) and a normal HAQ-DI score (≤0.5), respectively.
In the open-label period, significant improvements from baseline were observed at all timepoints in DAS28-ESR [mean change from baseline at week 24 (SD), −3.2 (1.4); p < 0.001], DAS28-CRP [−3.0 (1.3); p < 0.001], SDAI [−30.6 (14.3); p < 0.001], and CDAI [−29.3 (13.7); p < 0.001]. Similarly, throughout this period, CRP levels were significantly improved [mean change from baseline at week 24, −14.5 (27.6); p < 0.001], as were HAQ-DI scores [−0.8 (0.7); p < 0.001].
Response and other efficacy outcomes in the double-blind period
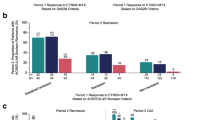
Among patients who met the criteria for LDA at the end of the open-label phase (week 24) and who were randomly assigned to one of the two treatment groups for the double-blind phase, a significantly higher proportion of patients in the etanercept combination-therapy group [71/163 (44%)] achieved the primary study endpoint of DAS28-ESR LDA at week 52 than patients in the placebo combination-therapy group [29/168 (17%); p < 0.001] (Fig. 2a). Significant differences were also seen between the etanercept and the placebo combination-therapy groups in the proportions of patients who had remission according to DAS28-ESR criteria, LDA and remission according to DAS28-CRP and SDAI, and LDA according to CDAI criteria at week 52 (Fig. 2a). In addition, similar results were observed for patients achieving ACR50 and ACR70 response criteria (Fig. 2b) and a normal HAQ-DI score (Fig. 2c).

Proportions of patients achieving a LDA and remission according to DAS28, SDAI, and CDAI criteria; b responses based on ACR criteria; and c minimal clinically important difference in HAQ-DI (>0.22) and normal HAQ-DI score (≤0.5) during the open-label (week 24) and double-blind (week 52) periods. Analyses conducted in the FAS population, using an LOCF approach. ACR American College of Rheumatology, CDAI clinical disease activity index, CRP C-reactive protein, DAS28 disease activity score in 28 joints, DAS28-CRP DAS 28 on the basis of CRP, DAS28-ESR DAS 28 on the basis of ESR, ESR erythrocyte sedimentation rate, FAS full analysis set, HAQ-DI Health Assessment Questionnaire-Disability Index, LDA low disease activity, LOCF last observation before rescue carried forward, SDAI simplified disease activity index. *p < 0.05, † p < 0.01, ‡ p < 0.001 between-group comparison for change from baseline
At weeks 28, 36, 44, and 52 in the double-blind period, the mean DAS28-ESR was significantly lower among patients receiving etanercept combination therapy than among those receiving placebo combination therapies (p < 0.001; Fig. 3a). A significant difference favoring biologic combination therapy was also observed in the DAS28-CRP, SDAI, and CDAI (Fig. 3a), as well as CRP concentrations (Fig. 3b) and HAQ-DI (Fig. 3c), at all timepoints during this period. Results for additional clinical and patient-reported outcomes during the double-blind period are summarized in Table 2.

Clinical and functional results: a DAS28-ESR, DAS28-CRP, SDAI, and CDAI, b CRP concentrations; and c HAQ-DI scores in patients receiving induction and maintenance therapy at week 0 and during the double-blind period. Analyses conducted in the FAS population, using an LOCF approach. CDAI clinical disease activity index, CRP C-reactive protein, DAS28 disease activity score in 28 joints, DAS28-CRP DAS 28 on the basis of CRP, DAS28-ESR DAS 28 on the basis of ESR, ESR erythrocyte sedimentation rate, FAS full analysis set, HAQ-DI Health Assessment Questionnaire-Disability Index, LOCF last observation before rescue carried forward, SDAI simplified disease activity index. *p < 0.05, † p < 0.01, ‡ p < 0.001 between-group comparison for change from baseline
Flare
In the double-blind period, 85 of 163 patients (52%) in the etanercept combination-therapy group and 134 of 168 patients (80%) in the placebo combination-therapy group experienced a flare (loss of LDA and worsening in DAS28-ESR ≥0.6 after week 24). The median time-to-flare (95% CI) in these groups was 197 days (141, 203) and 84 days (32, 85), respectively, in this period (p < 0.001; Supplementary Figure).
Sustained response
In the etanercept–methotrexate group, 38 of 67 patients (57%) who had DAS28 LDA at week 16 also achieved the primary endpoint (i.e., DAS28 LDA without rescue medication) at week 52, compared with 33 of 96 (34%) of those who did not have DAS28 LDA at week 16 (p = 0.005). In the placebo–methotrexate group, 20 of 71 (28%) patients who had DAS28 LDA at week 16 achieved the primary endpoint at week 52, versus 9 of 97 (9%) who did not have an early response (p = 0.001). However, the proportion of patients who achieved DAS28 LDA at week 52 was significantly higher in the etanercept–methotrexate group than in the placebo–methotrexate group in both those achieving and those not achieving week-16 LDA (p < 0.001).
Safety
A summary of safety findings in the open-label and double-blind periods is provided in Table 3. Adverse events were reported in 51% of patients in the open-label phase, and in 37 and 43% of the patients in the etanercept combination-therapy group and the placebo combination-therapy group, respectively, in the double-blind phase. One death occurred in the open-label period and none in the double-blind period. A 69-year-old Asian woman who had been receiving etanercept–methotrexate induction therapy and whose medical history included hyperthyroidism, angina pectoris, hypertension, anemia, and dizziness, died suddenly. Because an autopsy was not performed, the cause of death was unknown; treatment could not be ruled out as a possible cause, and the death was assessed as being related to treatment.
Treatment-related nonfatal serious adverse events occurring in the open-label period included herpes zoster, pneumonia, lower respiratory tract infection, salmonella sepsis, extradural abscess, and intervertebral discitis (n = 1, each); in the double-blind period, such events included sinusitis, urinary tract infection, and pneumonia [n = 1, each (all in the placebo combination-therapy group)]. No new safety signals were reported in either period of the study.
Discussion
In this two-period study conducted in Central and Eastern Europe, Latin America, the Middle East, Africa, and Asia, induction therapy with etanercept plus methotrexate, with or without other csDMARDs, following a T2T approach was effective in patients with long-standing RA and high disease activity at baseline. In the maintenance phase of the study, after induction of response, the etanercept combination regimen was significantly more effective in maintaining LDA and remission than a biologic-free regimen. Moreover, significantly higher proportions of patients receiving biologic combination therapy in the double-blind period achieved other clinical (e.g., ACR50 and ACR70 responses) and functional (e.g., normal HAQ-DI scores) endpoints. The safety profile of etanercept in this study was consistent with that observed in the previous clinical trials of the biologic in RA, with no unexpected safety issues observed in either the induction or maintenance study period.
Although this study was designed to evaluate the efficacy and safety of a T2T approach in patients with moderate-to-severe RA, the population evaluated had quite severe disease of long duration. The severity of disease in this patient population likely contributed to the high frequency of flare observed in the maintenance period, although the frequency of flare was lower in the biologic combination-therapy group than in the biologic-free combination-therapy group (52 vs. 80%, respectively). Patients in the etanercept group also experienced disease flare at a later timepoint than those in the placebo group in the double-blind period (197 vs. 84 days).
Improvements in the control of inflammation in RA with csDMARDs and biologics have resulted in markedly better clinical outcomes over the past few decades. A proportion of patients treated with these agents are able to achieve sustained remission, prompting questions about the appropriate management of this patient subgroup. Tapering or discontinuation of csDMARD or biologic therapy in patients who have maintained disease control over time is an attractive option, largely due to reduced treatment-related adverse events and costs and the desire to avoid needless treatment. However, the findings of this T2T study support continuation of biologic combination therapy in patients with an initial response, as it provided greater clinical and functional benefits than biologic-free combination therapy. For patients with established RA who achieve LDA, such as those in the present study, the ACR currently recommends continuation of csDMARDs and biologics, because cessation of treatment is unsuccessful in most patients [3]. Updated recommendations from the European League Against Rheumatism also cite the risk of flare upon withdrawal of biologic therapy in patients with established RA, and suggest that biologic tapering be considered only in those who have achieved persistent remission [9]. Similarly, based on available evidence, tapering of biologics is only recommended by the Asia Pacific League of Associations for Rheumatology in patients who have achieved clinical remission for at least 12 months [10].
The limitations of this study include the open-label study design in the induction period. Greater than 80% of patients enrolled in this study had severe disease activity, and the population had long-standing disease, which may explain in part the relatively low proportion of patients who achieved LDA at the end of the open-label period. The one-time assessment for randomization eligibility may have contributed to the high rates of disease flare observed in the double-blind phase (i.e., 52% and 80% of patients in the etanercept and placebo combination groups, respectively). A longer period of sustained LDA or remission may be necessary to confirm the treatment efficacy. In addition, of the initial 489 patients who received induction therapy in the open-label period, only 343 were treated in the randomized double-blind period and 316 completed the latter period. Approximately one-third of the population was, therefore, lost for evaluation, which may have affected the results. Moreover, in the double-blind treatment period, patients who achieved LDA at week 24 could be randomized to etanercept or placebo combination therapy, which may have introduced bias in the assessment of disease activity at the pre-randomization visit. A longer duration of induction combination therapy, resulting in a longer period of sustained LDA or remission, may have reduced the high prevalence of flare in the maintenance period. Moreover, a longer period of observation in the maintenance period among patients with this life-long disease would be necessary to determine the proportion who can achieve a sustained response in the biologic and biologic-free treatment groups. The absence of a randomized reduced-dose etanercept combination therapy group in the double-blind period may also be considered a limitation of the study design. In previous studies, biologic step-down therapy has been shown to more effectively maintain response than biologic withdrawal in patient populations with long-standing moderate RA [11] and with early moderate-to-severe disease [12]. In addition, no analyses were conducted to detect potential differences in response among patients in the etanercept and placebo combination-therapy groups associated with the various concomitant csDMARDs administered. Finally, patients enrolled in this study were not evaluated radiographically, precluding evaluation of the subclinical effects of a T2T approach.
In summary, the findings of this T2T study support the use of etanercept combination therapy as induction therapy in patients with moderate-to-severe RA and suggest that continuation of the biologic regimen gives results superior to a biologic-free regimen in those who achieve an initial response. Further research may be warranted to evaluate whether continuing a biologic regimen at a reduced dose would also result in superior disease control compared with a biologic-free regimen in patients with long-standing severe disease who have achieved response with full-dose biologic therapy.
References
van der Heijde DM, van Riel PL, van Leeuwen MA, van't Hof MA, van Rijswijk MH, van de Putte LB (1991) Older versus younger onset rheumatoid arthritis: results at onset and after 2 years of a prospective followup study of early rheumatoid arthritis. J Rheumatol 18:1285–1289
Plant MJ, Jones PW, Saklatvala J, Ollier WE, Dawes PT (1998) Patterns of radiological progression in early rheumatoid arthritis: results of an 8 year prospective study. J Rheumatol 25:417–426
Singh JA, Saag KG, Bridges SL Jr, Akl EA, Bannuru RR, Sullivan MC, Vaysbrot E, McNaughton C, Osani M, Shmerling RH, Curtis JR, Furst DE et al (2016) 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol 68:1–26. doi:10.1002/art.39480
Breedveld FC, Weisman MH, Kavanaugh AF, Cohen SB, Pavelka K, van Vollenhoven R, Sharp J, Perez JL, Spencer-Green GT (2006) The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum 54:26–37. doi:10.1002/art.21519
Emery P, Breedveld F, van der Heijde D, Ferraccioli G, Dougados M, Robertson D, Pedersen R, Koenig AS, Freundlich B (2010) Two-year clinical and radiographic results with combination etanercept-methotrexate therapy versus monotherapy in early rheumatoid arthritis: a two-year, double-blind, randomized study. Arthritis Rheum 62:674–682. doi:10.1002/art.27268
Kekow J, Moots RJ, Emery P, Durez P, Koenig A, Singh A, Pedersen R, Robertson D, Freundlich B, Sato R (2010) Patient-reported outcomes improve with etanercept plus methotrexate in active early rheumatoid arthritis and the improvement is strongly associated with remission: the COMET trial. Ann Rheum Dis 69:222–225. doi:10.1136/ard.2008.102509
Smolen JS, Han C, van der Heijde DM, Emery P, Bathon JM, Keystone E, Maini RN, Kalden JR, Aletaha D, Baker D, Han J, Bala M et al (2009) Radiographic changes in rheumatoid arthritis patients attaining different disease activity states with methotrexate monotherapy and infliximab plus methotrexate: the impacts of remission and tumour necrosis factor blockade. Ann Rheum Dis 68:823–827. doi:10.1136/ard.2008.090019
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS et al (1988) The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31:315–324
Smolen JS, Landewe R, Breedveld FC, Buch M, Burmester G, Dougados M, Emery P, Gaujoux-Viala C, Gossec L, Nam J, Ramiro S, Winthrop K et al (2014) EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis 73:492–509. doi:10.1136/annrheumdis-2013-204573
Lau CS, Chia F, Harrison A, Hsieh TY, Jain R, Jung SM, Kishimoto M, Kumar A, Leong KP, Li Z, Lichauco JJ, Louthrenoo W et al (2015) APLAR rheumatoid arthritis treatment recommendations. Int J Rheum Dis 18:685–713. doi:10.1111/1756-185X.12754
Smolen JS, Nash P, Durez P, Hall S, Ilivanova E, Irazoque-Palazuelos F, Miranda P, Park MC, Pavelka K, Pedersen R, Szumski A, Hammond C et al (2013) Maintenance, reduction, or withdrawal of etanercept after treatment with etanercept and methotrexate in patients with moderate rheumatoid arthritis (PRESERVE): a randomised controlled trial. Lancet 381:918–929. doi:10.1016/S0140-6736(12)61811-X
Emery P, Hammoudeh M, FitzGerald O, Combe B, Martin-Mola E, Buch MH, Krogulec M, Williams T, Gaylord S, Pedersen R, Bukowski J, Vlahos B (2014) Sustained remission with etanercept tapering in early rheumatoid arthritis. N Engl J Med 371:1781–1792. doi:10.1056/NEJMoa1316133
Acknowledgements
We wish to thank all patients who participated in the trial and all investigators and medical staff of all participating centers.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Karel Pavelka has received honoraria for lectures and consultancies from Amgen, AbbVie, Roche, Bristol-Myers Squibb (BMS), Merck Sharp & Dohme (MSD), and Pfizer. Nurullah Akkoç has received honoraria from and is an advisory board member for Pfizer, AbbVie, MSD, UCB, Novartis, and Amgen; research funding from Pfizer and UCB; and other grants from MSD. Cristiano Zerbini has received research grants from Novartis, Pfizer, BMS, Lilly, Amgen, Sanofi, and MSD; consulting fees from Pfizer, BMS, Lilly, and MSD; and is an advisory board member for Pfizer, Lilly, and Sanofi. Dmitry E. Karateev has received consulting and speakers’ bureau fees from AbbVie, BMS, Egis, Medac, MSD, Novartis, Pfizer, and Roche. Mustafa Al-Maini and Evgeny L. Nasonov declare no conflicts of interest. Mahboob Rahman was an employee of Pfizer during the conception/conduct of the study and owns Pfizer stock. Ron Pedersen, Andrew Dinh, Qi Shen, Radu Vasilescu, Sameer Kotak, Ehab Mahgoub, and Bonnie Vlahos are employees of Pfizer and Pfizer stockholders.
Funding
This study was funded by Pfizer. Editorial/medical writing support was provided by Donna McGuire of Engage Scientific Solutions and was funded by Pfizer.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pavelka, K., Akkoç, N., Al-Maini, M. et al. Maintenance of remission with combination etanercept–DMARD therapy versus DMARDs alone in active rheumatoid arthritis: results of an international treat-to-target study conducted in regions with limited biologic access. Rheumatol Int 37, 1469–1479 (2017). https://doi.org/10.1007/s00296-017-3749-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-017-3749-7