
Abstract
Behçet disease (BD) is a chronic relapsing multisystem disorder of unknown etiology, which preferentially affects the oral and genital mucous membranes, skin, and eyes. Neurological involvement is one of the most serious manifestations of BD, known as neuro-Behçet disease (NBD). We here describe clinical, radiological, and neuropathological findings for two patients with a possible variant of NBD, who manifested progressive ataxia in the absence of mucocutaneo-ocular signs characteristic for BD. Both patients presented a slowly progressive cerebellar phenotype, accompanied by behavioral changes and sphincter disturbance. Brain MRI scan revealed mild atrophy in pons and cerebellum. Both patients showed a mild CSF pleocytosis, and were positive for HLA-B51. The post-mortem examination performed in one patient, showed widespread foci of chronic encephalitis, consistent with the diagnosis of NBD. Steroid pulse therapy was effective in one patient. Identifying the progressive ataxia phenotype of NBD without mucocutaneo-ocular symptoms is important, because these patients may benefit from early steroid therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Behçet disease (BD) is a chronic relapsing multisystem disorder of unknown etiology, which preferentially affects the oral and genital mucous membranes, skin, and eyes [1]. Its prevalence is high in Japan, Middle East, and Mediterranean countries and has been linked with the human leucocyte antigen (HLA-B51) [2]. Although the prevalence of BD in Western countries was reported to be low, the disease could be underestimated [3]. Neurological involvement is one of the most serious manifestations of BD, affecting the quality of life, and was described in approximately 10% of cases with BD, known as neuro-Behçet disease (NBD) [4, 5]. We here describe clinical, radiological, and neuropathological findings for two patients with NBD, who manifested progressive ataxia in the absence of mucocutaneous-ocular signs characteristic for BD.
Case reports
Patient 1
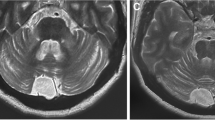
A 50-year-old male developed unsteadiness of gait at age 47. As his cerebellar symptoms slowly progressed, idiopathic late onset cerebellar ataxia (ILOCA) was initially suspected. At age 49 he noticed dysarthria and urinary incontinence, thus he was admitted for further examinations. On admission, he was emotionally labile and mentally disturbed (WAIS-R: total IQ, 84; VIQ, 81; and PIQ, 84). Neurological examinations revealed truncal ataxia, hyperreflexia, and positive Babinski signs. Urological examination revealed the presence of neurogenic bladder. He had not complained of headache or arthralgia. There were no signs of phlebitis, and pathergy test was negative. Routine blood and urinary examinations showed normal findings. Serum IgD and thyroid hormones were normal. Antinuclear antibody (ANA) and human immunodeficiency virus (HIV) antibody were negative. The cerebrospinal fluid (CSF) contained normal levels of protein (52 mg/dl) and IgG (6.8 mg/dl), but a slightly increased cell count of 20 μl−1. Oligoclonal band was negative. Brain MRI at age 49 revealed hyperintense lesions involving central pons (Fig. 1a), the slight atrophy of the pons and cerebellum (Fig. 1b), and the dilatation of ventricle III (Fig. 1c). Gene analysis revealed the absence of mutations for spinocerebellar ataxia (SCA) 1, 2, 6, Machado–Joseph disease (MJD), or dentatorubral-pallidoluysian atrophy (DRPLA). He was positive for HLA-B51 (A24, A31, B54, and CW1). Although mucocutaneo-ocular symptoms and signs were not evident, we diagnosed him as having NBD, based on the clinical features, CSF pleocytosis, MRI findings, and HLA typing. Prednisolone 40 mg/day was orally administered, but provided no significant benefits. The patient died from respiratory failure at age 50.

Brain MRI of Patient 1 (a–c) and Patient 2 (d–f). An axial T2-weighted image (a, TR 2,500, TE 100) shows hyperintense lesion involving the central pontine area. A T1-weighted image (TR 460, TE 25) shows a mild atrophy of the pons and cerebellum (b) and the dilatation of ventricle III (c). An axial T2-weighted image (d, TR 3,600, TE 96) and a T1-weighted image (e, TR 460, TE 9) reveal a mild atrophy of the pons and cerebellum. The dilatation of ventricle III is observed (f)
Neuropathological findings
The brain weighed 1,275 g and was atrophic with ventricular dilatation. Microscopically, there was a widespread multiple solitary and confluent foci of minute, recent to old necrotic lesions (Fig. 2a) in the central nervous system (CNS), including the cerebellum (Fig. 2b), cerebral white matter (Fig. 2c), pons (Fig. 2d), basal ganglia, thalamus, hypothalamus, optic nerve, and spinal cord. The recent necrotic lesions showed infiltration of lymphocytes, granulocytes, and macrophages. The presence of activated microglias and perivascular lymphocytic cuffing was also evident. The old lesions showed a marked fibrillary gliosis. These pathological findings were consistent with those of NBD.

Neuropathological findings of Patient 1. a Distribution of the pathological changes in the CNS. There is an extensive distribution of NBD lesions involving the pons, cerebellum, basal ganglia, and cerebral white matter. b A diffuse proliferation of lymphocytes, perivascular lymphocytic infiltration and marked gliosis are noted in the cerebellar dentate nucleus. H & E stain, bar: 100 μm. c A foci of the macrophage infiltration with gliosis observed in the cerebral white matter. H & E stain, bar: 50 μm. d Mild neuronal loss with fibrillary gliosis is noted in the pontine base. H & E stain, bar: 20 μm. e A binucleated neuron in the pontine nucleus. H & E stain, bar 10 μm
Patient 2
A 41-year-old male noticed unsteadiness of gait at age 39, which has gradually progressed. He has a past history of epididymitis. He subsequently noticed dysuria and speech disturbance, and visited our hospital. Brain MRI revealed a mild atrophy of the pons and cerebellum (Fig. 1d, e) as well as the dilatation of ventricle III (Fig. 1f). Initially the diagnosis of ILOCA was suspected. Gene analysis for SCAs (SCA1, 2, 6, MJD, and DRPLA) were all negative. As his neurological symptoms gradually worsened, he was admitted to our hospital at age 40. On admission, his mental state was slightly impaired (WAIS-R: total IQ, 79; VIQ, 84; and PIQ, 77), and a disinhibited personality was noted. Neurological examinations revealed dysarthria, truncal and limb ataxia, hyperreflexia, and positivity for the Babinski sign. The international cooperative ataxia rating scale (ICARS) was 33/100. He complained of pollakisuria and constipation. There were no signs of arthritis or phlebitis. Pathergy test was negative. Ophthalmologic examinations were normal. Routine blood and urinary examinations showed normal findings. Serum IgD and thyroid hormones were normal. Serum examinations were negative for ANA, HIV antibody or anti-neural antibodies (anti-Hu or anti-Yo). CSF contained a mildly elevated cell count of 12 μl−1, an increased level of β2-microglobulin (3,243 ng/ml) and a marked increase in the level of interleukin-6 (IL-6, 90.6 pg/ml), but normal protein and IgG levels. Oligoclonal band was negative. He was positive for HLA-B51 (A11, A24, B62, and CW3). On 3 Tesla proton MR spectroscopy (1H-MRS), there were reductions in N-acetyl-aspartate (NAA)/phosphocreatine (Cr) ratios of 1.55 and 1.48 in the pons (Fig. 3b) and cerebral white matter (Fig. 3e), respectively, whereas choline (Cho)/Cr ratio was not affected.

Single-voxel 1H-MRS was performed on Patient 2. The spectra were obtained using the point-resolved spectroscopy (PRESS) sequence combined with the chemical shift-selective excitation (CHESS) sequence to suppress the water signal (TR 2,000 ms, TE 80 ms). We placed 15 × 15 × 15 mm volume of interest in the pons (a) and parietal cerebral white matter (d). Decreased of NAA/Cr ratios of 1.55 and 1.48 in the pons (b) and cerebral white matter (e), respectively, were observed, whereas Cho/Cr ratio was not affected before steroid therapy. The NAA/Cr ratios in the pons (c) and cerebral white matter (f) improved to 1.65 and 1.93, respectively. The normal NAA/Cr and Cho/Cr ratios are indicated at the bottom as average ± SD
On the basis of the above findings, we made the diagnosis of NBD. Methylprednisolone pulse therapy (1 g/day) was started, followed by oral prednisolone at 60 mg/day. His neurological symptoms substantially improved as determined by the improvement of the ICARS score from 33/100 to 15/100. There were also marked improvement in the levels of CSF β2-microglobulin (from 3,243 to 2,000 ng/ml) and IL-6 (from 90.6 to 7.9 pg/ml). Moreover, MRS after steroid therapy, showed an improvement in NAA/Cr ratio in the pons (Fig. 3c) and cerebral white matter (Fig. 3f). The dose of oral prednisolone was gradually decreased; his neurological symptoms have remained stable.
Discussion
We here described two patients with peculiar clinical, radiological and neuropathological presentations, who were ultimately determined to represent a variant of NBD. These two patients presented progressive cerebellar signs accompanied by hyperreflexia, for which the initial diagnosis of ILOCA was made. Mild CSF pleocytosis as well as characteristic clinical features including behavioral changes and sphincter disturbance directed our attention to the diagnosis of NBD. The presence of HLA-B51 supports the diagnosis of NBD. Because little evidence of systemic manifestations of BD, including oral aphthae, genital ulcerations, skin lesions, eye involvement, or positive results of the pathergy test, was noted, the international diagnostic criteria of BD were not fulfilled in both cases [6].
NBD was reported to occur commonly several years after the onset of BD [4], and develops as an initial symptom in only 3% of cases. In our patients, the initial symptoms of cerebellar dysfunctions insidiously developed, followed by a slowly progressive course. Although the primary progressive phenotype was reported in 6 out of 42 cases of NBD [7], ataxia as a cardinal clinical feature of NBD has been only rarely reported. Hirohata et al. reported that the cases of 11 Japanese patients with progressive NBD, in which an elevated CSF IL-6 level and high prevalence (9/11) of HLA-B51 were observed [8]. An elevated CSF IL-6 level and the presence of HLA-B51 in our cases were consistent with their results.
The post-mortem examination was performed in Patient 1, and showed widespread foci of chronic encephalitis. The histopathological findings of NBD were typically chronic meningoencephalitis with scattered, focal, destructive lesions in the parenchyma, accompanied by perivascular lymphocytic infiltration [9]. It should be noted that although MRI could only detect brainstem and cerebellar lesions in Patient 1, widespread pathological changes involving the basal ganglia and cerebral white matter were also evident. Necropsies of five patients that developed clinically brainstem encephalitis in previous reports [10, 11] are of particular interest, because the neuropathological findings were very similar to those of Patient 1. Even though their neuropathological findings shared common features with those of NBD, in these previous reports, the clinical diagnosis of NBD was not made because mucocutaneo-ocular symptoms were not evident. Lueck et al. reported a case of chronic inflammatory disease of the CNS with uveitis, but at no time did he suffer from oral or genital ulceration or arthritis [12]. The neuropathological examination revealed histological changes compatible with a diagnosis of NBD [12]. Thus NBD without mucocutaneo-ocular symptoms may affect more patients than reported since it could be underdiagnosed.
MRI is useful in detecting CNS lesions in NBD [7]. The lesions are frequently detected in the brainstem, basal ganglia, or the deep white matter [7]. The prominent MRI finding in our cases was the dilatation of ventricle III, which has not been previously paid much attention. In Patient 2, MRS showed a decreased NAA/Cr ratio not only in the pons but also the in cerebral white matter, which suggests the widespread neuronal damage by NBD, even though conventional MRI failed to detect cerebral lesions. These abnormalities markedly improved after steroid therapy, suggesting that these changes could reflect disease activity. A previous report described a similar reduction in NAA/Cr ratio during the acute phase in three patients with NBD [13].
In conclusion, this report describes a possible variant of NBD manifesting a unique phenotype of progressive ataxia, which could be underdiagnosed because of the absence of mucocutaneo-ocular symptoms. It has been mentioned that BD may present with neurological symptoms before the ulceration becomes apparent [6, 14]. Identifying this peculiar phenotype of NBD by CSF examination, brain MRI, and HLA typing is important, because corticosteroid therapy could be effective if started early.
References
Sakane T, Takeno M, Suzuki N, Inaba G (1999) Behçet disease. N Engl J Med 341:1284–1291
Mizuki N, Inoko H, Ohno S (1997) Pathogenic gene responsible for the predisposition to Behçet’s disease. Int Rev Immunol 14:33–48
O’Duffy JD (1990) Behçet’s syndrome. N Engl J Med 322:326–327
Akman-Demir G, Serdaroglu P, Tasci B, The Neuro-Behçet Study Group (1999) Clinical patterns of neurological involvement in Behçet disease: evaluation of 200 patients. Brain 122:2171–2181
Kidd D, Steuer A, Denman AM, Rudge P (1999) Neurological complications in Behçet’s syndrome. Brain 122:2183–2194
International Study Group for Behçet’s Disease(1990) Criteria for diagnosis of Behçet disease. Lancet 335:1078–1080
Akman-Demir G, Bahar S, Coban O, Tasci B, Serdaroglu P (2003) Cranial MRI in Behçet’s disease: 134 examinations of 98 patients. Neuroradiology 45:851–859
Hirohata S, Issiki K, Oguchi H et al (1997) Cerebrospinal fluid interleukin-6 in progressive neuro-Behçet’s syndrome. Clin Immunol Immunopathol 82:12–17
Hadfield MG, Aydin HR, Lippman HR, Sanders KM (1997) Neuro-Behçet’s disease. Clin Neuropathol 16:55–60
Ueno T, Takahata N (1978) Chronic brainstem encephalitis with mental symptoms and ataxia. Report of three cases with necropsy. J Neurol Neurosurg Psychiatr 41:516–524
Iseki E, Iwabuchi K, Yagishita S, Amano N, Matsushita M (1988) Two necropsy cases of chronic encephalomyelitis: variants of neuro-Behçet’s disease. J Neurol Neurosurg Psychiatr 51:1084–1087
Lueck CJ, Pires M, McCartney ACE, Graham EM (1993) Ocular and neurological Behçet disease without orogenital ulceration? J Neurol Neurosurg Psychiatr 56:505–508
Nüssel F, Wegmüller H, Laseyras F, Posse S, Herschkowits N, Huber P (1991) Neuro-Behçet: acute and sequential aspects by MRI and MRS. Eur Neurol 31:399–402
Sigal LH (1987) The neurologic presentation of vasculitic and rheumatologic syndromes. Medicine 66:157–180
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hirose, M., Ikeuchi, T., Hayashi, S. et al. A possible variant of neuro-Behçet disease presenting chronic progressive ataxia without mucocutaneo-ocular symptoms. Rheumatol Int 27, 61–65 (2006). https://doi.org/10.1007/s00296-006-0171-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-006-0171-y