
Abstract
Constitutive heterochromatin packages long stretches of repetitive DNA sequences at the centromere and telomere, and ensures genomic integrity at these loci by preventing aberrant recombination and transcription. The chromatin scaffold of heterochromatin is dynamically regulated in the cell cycle, and inheritance of the epigenetically silenced state is dependent on a transcriptional event imposed on the underlying non-coding RNA in conjunction with the DNA replicative phase. Heterochromatin becomes transiently loosened in response to a reduction in the binding of Swi6, a heterochromatin protein, and this allows RNA polymerase II access to the underlying sequence. The derived transcripts, in turn, drive heterochromatin formation via the recruitment of other silencing factors. It remains unclear how heterochromatin becomes decompacted in a cell cycle-specific manner. Here, we describe a mechanism of heterochromatin decompaction initiated by a novel histone modification, histone H3 tyrosine 41 phosphorylation (H3Y41p). We will discuss how H3Y41p cooperates with other regulatory pathways to enforce cell cycle-dependent regulation of constitutive heterochromatin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The DNA double helix of eukaryotic cells is condensed and organized around scaffolding histone proteins, referred to as chromatin. The basic subunit of chromatin is the nucleosome, which is composed of octameric histone proteins (two copies each of histone H2A, H2B, H3 and H4) complexed with 1.6 turns of DNA (~ 146 base pairs [bp]) (Luger et al. 1997). The degree of chromatin compaction at different chromosomal loci directly affects its ability to be transcribed. Chromatin is, therefore, roughly classified as euchromatin, which is relatively opened and common to most coding sequences, and heterochromatin, which is more compacted and typical of highly repetitive regions. Euchromatin is preferentially enriched with histone modifications associated with transcriptional activation, such as histone H3 lysine 4 and 36 methylation (H3K4me and H3K36me, respectively), whereas heterochromatin is nested with transcriptionally repressive modifications, such as H3K9me and H3K27me (Cam et al. 2005; Shilatifard 2006; Simon and Kingston 2009). Heterochromatin is further subdivided into facultative and constitutive heterochromatin, with the former common to regions of DNA that code for developmental genes, retaining the potential to be transcriptionally activated in response to environmental cues (Trojer and Reinberg 2007; Seo et al. 2018). Constitutive heterochromatin, on the other hand, generally encompasses non-coding repetitive DNA associated with specialized chromosomal loci, such as centromeres and telomeres (Allshire and Ekwall 2015).
Much research in the past 2 decades has mechanistically defined H3K9me as the “binding site” recognized by various protein motifs during heterochromatin formation, particularly proteins that contain chromodomains (CDs) (Bannister et al. 2001; Nakayama et al. 2001; Sadaie et al. 2008). CDs are protein structural domains of approximately 40–50 amino acids in length that are typically found within proteins of the CD superfamily, founded by mammalian heterochromatin protein 1 (HP1). HP1 proteins assemble various macromolecular complexes on constitutive heterochromatin that act as platforms for the recruitment of other protein complexes with chromatin-modifying activities (Bannister et al. 2001; Grewal and Jia 2007; Cam et al. 2009). Together with di- and trimethylated H3K9 (H3K9me2 and H3K9me3, respectively), HP1 proteins regulate transcriptional silencing in all eukaryotic organisms, except certain ascomycetes fungi, such as the budding yeast Saccharomyces cerevisiae (Lomberk et al. 2006).
Heterochromatin in fission yeast, however, shares structural similarities with that of other eukaryotes, and is, therefore, widely employed as a model organism to study heterochromatin formation. For example, fission yeast encompasses long stretches of repetitive elements termed dg and dh, which are similar to the tandem α-satellite repeats found in human pericentromeric heterochromatin (Bjerling and Ekwall 2002; Verdel and Moazed 2005; Yang and Li 2017). The human genome encodes three HP1 proteins—HP1α, HP1β and HP1γ (Lomberk et al. 2006)—represented as Swi6 and Chp2 in fission yeast (Thon and Verhein-Hansen 2000). Fission yeast also contains Chp1 and Clr4, two other CD-containing proteins that recognize H3K9me2/3 and are important for heterochromatin formation: Chp1 is a subunit of the RNA-induced transcriptional silencing (RITS) complex, whereas Clr4 is the catalytic subunit of the H3K9 methyltransferase complex, Clr-C (Verdel et al. 2004; Sadaie et al. 2004; Zhang et al. 2008). These CD proteins interact with a host of heterochromatin-regulating factors to establish and maintain heterochromatin throughout the cell cycle in fission yeast.
Dynamic regulation of centromeric heterochromatin in cell cycle
Proper establishment and maintenance of heterochromatin are critical for ensuring genomic integrity. Disruptions to heterochromatin silencing often underlie defects in mitotic and meiotic chromosomal segregation, hypersensitivity towards DNA damaging agents, and abnormal recombination events that lead to DNA sequence loss and genomic instability (Ekwall et al. 1996; Kato et al. 2005; Li et al. 2013). Constitutive heterochromatin, therefore, remains in a condensed form to restrict the accessibility of DNA processing factors (Nicolas et al. 2007; Meschini et al. 2015) and prevents aberrant transcription of heterochromatic repeats. Indeed, such events are a hallmark of heterochromatin disruption (for examples, see Bayne et al. 2008; Chen et al. 2008; Kitano et al. 2011). However, recent evidence suggests a role for RNA polymerase II-mediated transcription as being important for perpetuating the silenced state of heterochromatin through dynamic regulation during the cell cycle (Cam et al. 2005; Kato et al. 2005; Djupedal et al. 2005).
There is a short window within the S-phase of the cell cycle where a preferential transcriptional “burst” coincides with the preferential binding of RNA polymerase II (Chen et al. 2008; Kloc et al. 2008). This burst of transcription was identified by quantifying the levels of non-coding transcripts from constitutive heterochromatin at centromeric and sub-telomeric regions, and mating-type locus. Transcripts arising from heterochromatic repeats are processed by RNA interference (RNAi) machineries into small interference RNA (siRNA), which becomes incorporated into the RITS complex, and direct the complex to cognate heterochromatic locations via base pairing with heterochromatic transcripts (Motamedi et al. 2004; Noma et al. 2004; Sugiyama et al. 2005). This occurs in conjunction with Clr-C, H3K36 methyltransferase Set2, and several histone deacetylase (HDAC) complexes including Class I and II complexes—with Clr6 and Clr3 as catalytic subunits, respectively―to reconstitute the condensed heterochromatin at the end of S-phase (Chen et al. 2008; Cam et al. 2009). The S-phase-related transcription of centromeric heterochromatic sequences is correlated with the temporal colocalization of factors and histone modifications associated with transcriptional activation, and also with colocalization of the DNA replicative polymerase ε complex, which likely coordinates the transmission of epigenetic silencing on newly synthesized DNA at the centromeric heterochromatin (Li et al. 2011).
Similar S-phase-linked heterochromatic transcription has been reported in mammalian cells; albeit, not in as much molecular detail as that in fission yeast. In mouse mammary epithelial cells, RNA polymerase II-dependent transcripts were detected from γ-satellites during the G1- to S-phase of the cell cycle (Lu and Gilbert 2007). In mouse embryonic stem cells, the developmentally controlled formation of heterochromatin upon retinoic acid induction is associated with HP1α and transcription from pericentromeric satellite DNA, concomitant with the binding of transcription regulatory p68 RNA-helicase (Enukashvily et al. 2009). Heterochromatic transcription also maintains murine heterochromatic chromocenter formation in later developmental stages (Probst et al. 2010). Importantly, these heterochromatic loci-originated transcripts serve important physiological functions in heterochromatin formation, for example, in the targeting of HP1α to heterochromatic loci (Maison et al. 2011). Transcripts of pericentromeric high-copy satellite repeats can sequester methyl-CpG-binding protein 2 (MeCP2) to form cancer-associated satellite transcript (CAST) bodies, which in turn repress the transcription of these repeats (Hall et al. 2017). Transcriptional silencing of heterochromatic repeats appears to perform a tumor-suppressing function as the de-repression of pericentromeric satellite transcription is associated with tumorigenesis (Eymery et al. 2009; Ting et al. 2011; Hall et al. 2017). Transcription and RNAi-coupled mechanisms can also silence centromeric retrotransposon repeats in plants (Neumann et al. 2007; Hale et al. 2016). This transcription-dependent mechanism is likely to be universal in regulating heterochromatin formation: it has also been reported in the silencing of murine telomeric repeats (Mallm and Rippe 2015). Taken together, the initiation of heterochromatin formation by the transcription of heterochromatin-associated non-coding repetitive DNA sequences represents a conserved mechanism to maintain the balance between silencing and transcription for precise epigenetic inheritance of silenced chromatin memory.
Molecular interactions that reconstitute heterochromatin associated with non-coding transcription
Several intermolecular interactions enable the reconstitution of silenced chromatin. For example, within the RITS complex, the Tas3 subunit acts as a scaffold joining Ago1 to Chp1, which contains a CD capable of binding to H3K9me2/3 (DeBeauchamp et al. 2008); this suggests a cooperation between RNAi and H3K9me in targeting RITS to pericentromeric heterochromatin (Zhang et al. 2008). Methylation of H3K9 by Clr4 in the Clr-C complex also directs other CD proteins—Swi6 and Chp2—to heterochromatin (Sadaie et al. 2008; Nakayama et al. 2001). Chp2, in turn, recruits a type II histone deacetylase snf2/HDAC-containing repressor complex (SHREC) to deacetylate chiefly histone H3 lysine 14 (Sugiyama et al. 2007; Motamedi et al. 2008). A type I HDAC complex is also localized to centromeric chromatin, where its catalytic subunit Clr6 confers pan-deacetylation at multiple lysine residues on histones of heterochromatic nucleosomes. H3K36me is transiently enriched at pericentromeric heterochromatin during the preferential localization of RNA polymerase II, whose C-terminus can recruit the Set2 H3K36 methyltransferase (Chen et al. 2008; Cam et al. 2009; Suzuki et al. 2016). Collectively, the deacetylation and methylation of histones coordinate with canonical heterochromatin-associated factors to establish and spread heterochromatin on newly synthesized DNA (Chen et al. 2008; Li et al. 2011; Zhang et al. 2008).
Modulation of centromeric heterochromatin for transcription by RNA polymerase II
Compaction of the heterochromatin scaffold must be dynamically adapted during different stages of the cell cycle, the most challenging of which is inarguably the DNA replication phase, in which the condensed chromatin needs to be loosened for the passage of DNA polymerases and the RNA polymerase to establish the silenced state. This loosening poses an obvious threat to genomic stability, as loosened chromatin can lead to an unraveling of the entire heterochromatin structure. Hence, there exist mechanisms that regulate the “opening up” of heterochromatin only during appropriate times in the cell cycle. We and others have reported the temporary loosening of heterochromatin after Swi6 delocalization, which is initiated at the previous M-phase (Chen et al. 2008; Kloc et al. 2008), and in mutants of heterochromatin-associated factors, such as Clr4 and Swi6 mutants, heterochromatin structure is abolished (Chen et al. 2008; Nakayama et al. 2001).
Histone H3 phosphorylation at serine 10 (H3S10p) is a mitotic event closely associated with chromosomal condensation in M-phase (Johansen and Johansen 2006; Chen et al. 2008). In fission yeast, factors constituting the chromosomal condensin complex localize to centromeric chromatin in a H3S10-dependent manner (Chen et al. 2008); these factors are also required for maintaining heterochromatic silencing at the centromere. The mechanism underlying the dynamic heterochromatin localization of the condensin complex is unclear but it may be regulated by non-structural maintenance of chromosome (SMC) subunits (Robellet et al. 2017). In mammalian cells, centromeric nucleosomes become phosphorylated on H3S10 during M-phase, which downregulates the binding of HP1α to H3K9me (Hirota et al. 2005). This points to a binary methyl/phospho switch, wherein modifications on H3K9/S10 coordinate heterochromatic silencing (Fischle et al. 2005; Hirota et al. 2005). However, H3K9me is also transiently decreased (even though not completely abolished) on heterochromatin from M-phase through to S-phase, exhibiting a preferential non-coding transcription (Chen et al. 2008), suggesting that other epigenetic mechanisms may modulate the chromatin scaffold at the centromere to elicit “opening up” of heterochromatin.
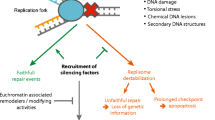
Recently, we discovered that phosphorylation of histone H3 at tyrosine 41 (H3Y41p) can prime heterochromatin for S-phase-dependent transcription of centromeric repeats. H3Y41p is tightly regulated during the cell cycle, with its phosphorylation in M-phase persisting to mid-S-phase; this occurs before the peak in the septation index is achieved, and its presence overlaps with Swi6 displacement from centromeric heterochromatin (Ren et al. 2018). In an in vitro binding assay, we showed that H3Y41p is capable of displacing H3K9me2-associated Swi6, and in the H3Y41F mutant, where all three copies of the histone H3 genes express phenylalanine (F) at position 41, the lack of H3Y41 promotes centromeric association of Swi6 in vivo (Ren et al. 2018). Our observation that the phosphorylation of H3Y41 excludes Swi6 is consistent with that observed in leukemic cancer cell lines overexpressing the JAK2 kinase. JAK2 phosphorylates H3Y41 at promoters of certain oncogenic genes, for example Imo2 ectopically, which, in turn, de-represses gene transcription via antagonizing HP1α binding (Dawson et al. 2009). However, further examination revealed that the effect of H3Y41p differs for different CD proteins. Unlike Swi6, H3Y41p does not affect the heterochromatic association of Chp2, another CD protein that performs a more minor role in centromeric heterochromatin silencing; albeit, this is probably due to the lower level of expression of Chp2 in vivo. Yet, H3Y41p enhances the binding of Chp1 to the histone H3 tail, and, in the H3Y41F mutant, this corresponds to a reduced association of Chp1 with centromeric heterochromatin. Therefore, we propose that H3Y41p safeguards the switch between Swi6 and Chp1, which “opens the gate” for RNA polymerase II to access heterochromatic repeats at the centromere during S-phase. Displacement of Swi6 unravels the compacted chromatin, and the RNAi machinery, including the RITS complex, is recruited to process RNA polymerase II-derived nascent heterochromatic transcripts (Fig. 1).

Model depicting coordination of the Swi6/Chp1 switch during the M- to S-phase of the cell cycle following H3Y41 phosphorylation
Reciprocally, histone H3 lysine 4 acetylation (H3K4ac) opposes the action of H3Y41p to revert the CD switch and re-establish Swi6 binding as the cell traverses from S- to G2-phase. H3K4ac also reduces the affinities of Chp1 and Clr4 for H3K9me and enhances the recruitment of Swi6 and Chp2 (Xhemalce and Kouzarides 2010), possibly “closing the gate” for RNA polymerase II access to the underlying heterochromatic sequences (Fig. 1). Even though both Swi6 and Chp2 belong to the HP1 family, they nevertheless differentially interact with histone modifications, indicating their distinct roles in heterochromatic silencing during S-phase of the cell cycle (Motamedi et al. 2008; Sadaie et al. 2008; Issac et al. 2017). Swi6 contributes to heterochromatin assembly through an auto-inhibition mechanism, undergoing a conformational change between an auto-inhibitory state and a spreading-competent state in response to changes in the interaction between Swi6 and the nucleosome. These conformational changes likely reflect the involvement of the ARKGGG motif in Swi6 that is not found in Chp2 (Canzio et al. 2013). Further differences may also arise from the differences in how Swi6 and Chp2 undergo homodimerization (Issac et al. 2017).
Perspectives on cell cycle-regulated heterochromatic transcription
Although many studies have advanced our knowledge on the dynamic assembly, spread, and maintenance of heterochromatin, how these mechanisms are integrated for the inheritance of epigenetic memory in conjunction with the cell cycle is still far from understood. Several of the studies discussed above have shown the central role of nascent non-coding transcripts derived from heterochromatic sequences as a point of convergence between transcriptional activation and repression. Even though cell cycle-regulated heterochromatic transcription has been observed in several organisms to silence repeats of widely varying sequences (refer above), the mechanistic details of the S-phase-specific localization of RNA polymerases have only been elucidated in fission yeast.
Our discovery that H3Y41p regulates the delocalization of Swi6 in the cell cycle suggests a conserved regulation of the heterochromatin scaffold, as H3Y41p also displaces HP1α to reverse transcriptional repression in human cells (Ren et al. 2018; Dawson et al. 2009). However, the observation that H3Y41p does not simply antagonize the binding of the CD protein (Swi6) but can, conversely, recruit another (Chp1) CD protein in fission yeast reveals the intricacy of H3Y41p-based regulation. This suggests that H3Y41p is not simply a “code” but that its signaling may combine with other mechanisms of repression to elicit specific effects on the switch between CD proteins. Indeed, previous work has pointed to the roles of RNA polymerase II and nascent transcripts in contributing to the recruitment of silencing factors during S-phase when heterochromatin is less compacted. For instance, the Rik1 component of the Clr-C complex physically interacts with RITS, which, in turn, mounts onto the nascent transcript (Chen et al. 2008; Cam et al. 2009). The phosphorylation of H3Y41 possibly serves to ensure the recruitment of RITS preferentially to heterochromatic loci to differentiate RNA polymerase II transcription at heterochromatin from that occurring at other regions of the genome during the critical S-phase window.
The use of a transcriptional event to induce silencing on chromatin is potentially risky. This is because transcriptional silencing, once initiated, is self-propagating and can spread over an extended distance. Indeed, mis-targeted heterochromatin formation on other euchromatic regions can result in aberrant silencing of long stretches of open reading frames to compromise cellular health (Ci et al. 2018; Salzberg et al. 2017). Thus, it is important for the cell to induce silenced chromatin formation only on pre-determined heterochromatic loci. To induce such specificity, it is possible for other histone modifications, besides H3Y41p and H3K4ac, to be involved. These histone marks may interplay to set up a combinatorial code to direct protein complexes with appropriate transcriptional modulating activities to the heterochromatin during the brief window of opportunity during S-phase. Consistent with this view, acetylation of histone H3 lysine 9 (H3K9ac) and lysine 14 (H3K14ac) can also regulate the localization of the RITS complex (Deng et al. 2015; Reddy et al. 2011). Furthermore, different levels of H3K9 methylation (di- or tri-methylation)—shown to differentially affect heterochromatic silencing (Jih et al. 2017; Yu et al. 2018)—can also modulate the degree to which silencing can be differentially downregulated in the cell cycle. These histone modifications may further cooperate with other transacting factors to epigenetically propagate the heterochromatin as a site for RNAPII loading during S-phase (D’Urso and Brickner 2017), and one such factor may be the H2B uniquitin ligase complex (HULC), which can promote RNAPII occupancy at centromeric heterochromatin (Zofall and Grewal 2007; Chen et al. 2008).
The phosphorylation of HP1α proteins in association with DNA leads to a physicochemical, phase-separated space, which is proposed to compartmentalize heterochromatin formation (Larson et al. 2017). If similar phase-separated localities are formed on Swi6-coated heterochromatin in fission yeast, it is possible that the partial depletion of Swi6 during the transition from M- to S-phase may result in an intermediately separated compartment—separable from the compacted interphase heterochromatin and unheterochromatized euchromatic regions—to facilitate the epigenetic inheritance of the silenced chromatin.
Defects in heterochromatin architecture that cause the de-repression of repetitive sequences are associated with tumorigenesis (Eymery et al. 2009; Ting et al. 2011; Hall et al. 2017). Thus, it is important to gain an understanding of the regulation of epigenetic inheritance as it pertains to heterochromatic integrity, which may lead to the identification of factors that can be employed as prognostic or diagnostic markers for cancer. Furthermore, it will be important to identify therapeutic targets that could be used to fine-tune genomic stability in combination with other anti-cancer therapies. Recently, a synthetic lethality approach that exploits on concurrently inactivating synergistically acting oncogenic pathways has been shown effective against many hitherto undruggable malignancies (Jackson and Chen 2016; Chen 2018). Heterochromatic disruption results in the accumulation of non-coding transcripts arising from the repetitive DNA elements, is causally linked to carcinogenesis in malignancies including ovary, lung and colon (Zhu et al. 2011; Ting et al. 2011). In fission yeast, centromere disruption—correlating with upregulated transcription of centromeric repetitive sequences—compromised tolerance against the topoisomerase II inhibitor doxorubicin, which is a widely used anti-cancer drug in the clinics (Nguyen et al. 2015, 2016). Thus, the modulation of H3Y41p level to impact oncogenic expression and centromeric integrity may be combined with chemotherapeutic agents to effect killing of cancer cells in a synthetic lethal manner (Jackson and Chen 2016; Chen 2018).
Concluding remarks
The phosphorylation of H3Y41 cooperates with other regulatory pathways to enforce cell cycle-specific loosening of centromeric heterochromatin. H3Y41p induces a switch between CD proteins: the displacement of Swi6/HP1 to provide RNA polymerase II with access to the DNA, and the recruitment of Chp1, a RITS component, for the RNAi-mediated establishment of heterochromatin. Hence, the conditioning of chromatin scaffolds through H3Y41 phosphorylation makes way for transcriptional silencing in S-phase to ensure the precise epigenetic propagation of silenced, repetitive DNA elements of heterochromatin.
References
Allshire RC, Ekwall K (2015) Epigenetic regulation of chromatin states in Schizosaccharomyces pombe. Cold Spring Harb Perspect Biol 7:a018770. https://doi.org/10.1101/cshperspect.a018770
Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410:120–124
Bayne EH, Portoso M, Kagansky A, Kos-Braun IC, Urano T, Ekwall K, Alves F, Rappsilber J, Allshire RC (2008) Splicing factors facilitate RNAi-directed silencing in fission yeast. Science 322:602–606. https://doi.org/10.1126/science.1164029
Bjerling P, Ekwall K (2002) Centromere domain organization and histone modifications. Braz J Med Biol Res 35:499–507
Cam HP, Sugiyama T, Chen ES, Chen X, FitzGerald PC, Grewal SI (2005) Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat Genet 37:809–819
Cam HP, Chen ES, Grewal SI (2009) Transcriptional scaffolds for heterochromatin assembly. Cell 136:610–614. https://doi.org/10.1016/j.cell.2009.02.004
Canzio D et al (2013) A conformational switch in HP1 releases auto-inhibition to drive heterochromatin assembly. Nature 496:377–381. https://doi.org/10.1038/nature12032
Chen ES (2018) Targeting epigenetics using synthetic lethality in precision medicine. Cell Mol Life Sci 75:3381–3392. https://doi.org/10.1007/s00018-018-2866-0
Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SI (2008) Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature 451:734–737. https://doi.org/10.1038/nature06561
Ci X et al (2018) Heterochromatin protein 1α mediates development and aggressiveness of neuroendocrine prostate cancer. Cancer Res 78:2691–2704. https://doi.org/10.1158/0008-5472.CAN-17-3677
D’Urso A, Brickner JH (2017) Epigenetic transcriptional memory. Curr Genet 63:435–439. https://doi.org/10.1007/s00294-016-0661-8
Dawson MA, Bannister AJ, Göttgens B, Foster SD, Bartke T, Green AR, Kouzarides T (2009) JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 461:819–822. https://doi.org/10.1038/nature08448
Debeauchamp JL, Moses A, Noffsinger VJ, Ulrich DL, Job G, Kosinski AM, Partridge JF (2008) Chp1-Tas3 interaction is required to recruit RITS to fission yeast centromeres and for maintenance of centromeric heterochromatin. Mol Cell Biol 28:2154–2166. https://doi.org/10.1128/MCB.01637-07
Deng X, Zhou H, Zheng G, Wang W, Mao L, Zhou X, Yu Y, Lu H (2015) Sgf73, a subunit of SAGA complex, is required for the assembly of RITS complex in fission yeast. Sci Rep 5:14707. https://doi.org/10.1038/srep14707
Djupedal I, Portoso M, Spåhr H, Bonilla C, Gustafsson CM, Allshire RC, Ekwall K (2005) RNA Pol II subunit Rpb7 promotes centromeric transcription and RNAi-directed chromatin silencing. Genes Dev 19:2301–2306
Ekwall K, Nimmo ER, Javerzat JP, Borgstrøm B, Egel R, Cranston G, Allshire R (1996) Mutations in the fission yeast silencing factors clr4+ and rik1+ disrupt the localisation of the chromo domain protein Swi6p and impair centromere function. J Cell Sci 109:2637–2648
Enukashvily NI, Malashicheva AB, Waisertreiger IS (2009) Satellite DNA spatial localization and transcriptional activity in mouse embryonic E-14 and IOUD2 stem cells. Cytogenet Genome Res 124:277–287. https://doi.org/10.1159/000218132
Eymery A et al (2009) A transcriptomic analysis of human centromeric and pericentric sequences in normal and tumor cells. Nucleic Acids Res 37:6340–6354. https://doi.org/10.1093/nar/gkp639
Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD (2005) Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438:1116–1122
Grewal SI, Jia S (2007) Heterochromatin revisited. Nat Rev Genet 8:35–46
Hale CJ, Potok ME, Lopez J, Do T, Liu A, Gallego-Bartolome J, Michaels SD, Jacobsen SE (2016) Identification of multiple proteins coupling transcriptional genes silencing to genome stability in Arabidopsis thaliana. PLoS Genet 12:e1006092. https://doi.org/10.1371/journal.pgen.1006092
Hall LL, Byron M, Carone DM, Whitfield TW, Pouliot GP, Fischer A, Jones P, Lawrence JB (2017) Demethylated HSATII DNA and HSATII RNA foci sequester PRC1 and MeCP2 into cancer-specific nuclear bodies. Cell Rep 18:2943–2956. https://doi.org/10.1016/j.celrep.2017.02.072
Hirota T, Lipp JJ, Toh BH, Peters JM (2005) Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 438:1176–1180
Issac RS, Sanulli S, Tibble R, Hornsby M, Ravalin M, Craik CS, Gross JD, Narlikar GJ (2017) Biochemical basis for distinct roles of the heterochromatin proteins Swi6 and Chp2. J Mol Biol 429:3666–3677. https://doi.org/10.1016/j.jmb.2017.09.012
Jackson RA, Chen ES (2016) Synthetic lethal approaches for assessing combinatorial efficacy of chemotherapeutic drugs. Pharmacol Ther 162:69–85. https://doi.org/10.1016/pharmthera.2016.01.014
Jih G, Iglesias N, Currie MA, Bhanu NV, Paulo JA, Gygi SP, Garcia BA, Moazed D (2017) Unique roles for histone H3K9me states in RNAi and heritable silencing of transcription. Nature 547:463–467. https://doi.org/10.1038/nature23267
Johansen KM, Johansen J (2006) Regulation of chromatin structure by histone H3S10 phosphorylation. Chromosome Res 14:393–404. https://doi.org/10.1007/s10577-006-1063-4
Kato H, Goto DB, Martienssen RA, Urano T, Furukawa K, Murakami Y (2005) RNA polymerase II is required for RNAi-dependent heterochromatin assembly. Science 309:467–469
Kitano E, Hayashi A, Kanai D, Shinmyozu K, Nakayama JI (2011) Roles of fission yeast Grc3 protein in ribosomal RNA processing and heterochromatic gene silencing. J Biol Chem 286:15391–15402. https://doi.org/10.1074/jbc.M110.201343
Kloc A, Zaratiegui M, Nora E, Martienssen R (2008) RNA interference guides histone modification during the S phase of chromosomal replication. Curr Biol 18:490–495. https://doi.org/10.1016/j.cub.2008.03.016
Larson AG, Elnatan D, Keenen MM, Tmka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, Narlikar GJ (2017) Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547:236–240. https://doi.org/10.1038/nature22822
Li F, Martienssen R, Cande WZ (2011) Coordination of DNA replication and histone modification by the Rik1-Dos2 complex. Nature 475:244–248. https://doi.org/10.1038/nature10161
Li PC, Petreaca RC, Jensen A, Yuan JP, Green MD, Forsburg SL (2013) Replication fork stability is essential for the maintenance of centromere integrity in the absence of heterochromatin. Cell Rep 3:638–645. https://doi.org/10.1016/j.celrep.2013.02.007
Lomberk G, Wallrath L, Urrutia R (2006) The heterochromatin protein 1 family. Genome Biol 7:228. https://doi.org/10.1186/gb-2006-7-7-228
Lu J, Gilbert DM (2007) Proliferation-dependent and cell cycle regulated transcription of mouse pericentric heterochromatin. J Cell Biol 179:411–421
Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260
Maison C et al (2011) SUMOylation promotes de novo targeting of HP1α to pericentric heterochromatin. Nat Genet 43:220–227. https://doi.org/10.1038/ng.765
Mallm JP, Rippe K (2015) Aurora kinase B regulates telomerase activity via a centromeric RNA in stem cells. Cell Rep 11:1667–1678. https://doi.org/10.1016/j.celrep.2015.05.015
Meschini R, Morucci E, Bemi A, Lopez-Martinez W, Palitti F (2015) Role of chromatin structure modulation by the histone deacetylase inhibitor trichostatin A on the radio-sensitivity of ataxia telangiectasia. Mutat Res 777:52–59. https://doi.org/10.1016/j.mrfmmm.2015.04.009
Motamedi MR, Verdel A, Colmenares SU, Gerber SA, Gygi SP, Moazed D (2004) Two RNAi complexes, RITS and RDRC, physically interact and localize to noncoding centromeric RNAs. Cell 119:789–802. https://doi.org/10.1016/j.cell.2004.11.034
Motamedi MR, Hong EJ, Li X, Gerber S, Denison C, Gygi S, Moazed D (2008) HP1 proteins form distinct complexes and mediate heterochromatic gene silencing by nonoverlapping mechanisms. Mol Cell 32:778–790. https://doi.org/10.1016/j.molcel.2008.10.026
Nakayama JI, Rice JC, Strahl BD, Allis CD, Grewal SI (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292:110–113
Neumann P, Yan H, Jiang J (2007) The centromeric retrotransposons of rice are transcribed and differentially processed by RNA interference. Genetics 176:749–761. https://doi.org/10.1534/genetics.107.071902
Nguyen TT, Lim JS, Tang RM, Zhang L, Chen ES (2015) Fitness profiling links topoisomerase II regulation of centromeric integrity to doxorubicin resistance in fission yeast. Sci Rep 5:8400. https://doi.org/10.1038/srep08400
Nguyen TT et al (2016) Predicting chemotherapeutic drug combinations through gene network profiling. Sci Rep 6:18658. https://doi.org/10.1038/srep18658
Nicolas E, Yamada T, Cam HP, Fitzgerald PC, Kobayashi R, Grewal SI (2007) Distinct roles of HDAC complexes in promoter silencing, antisense suppression and DNA damage protection. Nat Struct Mol Biol 14:372–380
Noma K, Sugiyama T, Cam H, Verdel A, Zofall M, Jia S, Moazed D, Grewal SI (2004) RITS acts in cis to promote RNA interference-mediated transcriptional and post-transcriptional silencing. Nat Genet 36:1174–1180
Probst AV, Okamoto I, Casanova M, El Marjou F, Le Baccon P, Almouzni G (2010) A strand-specific burst in transcription of pericentric satellites is required for chromocenter formation in early mouse development. Dev Cell 19:625–638. https://doi.org/10.1016/j.devcel.2010.09.002
Reddy BD, Wang Y, Niu L, Higuchi RC, Marguerat SB, Bähler J, Smith GR, Jia S (2011) Elimination of a specific histone H3K14 acetyltransferase complex bypasses the RNAi pathway to regulate pericentric heterochromatin functions. Genes Dev 25:214–219. https://doi.org/10.1101/gad.1993611
Ren B, Tan HL, Nguyen TTT, Sayed AMM, Li Y, Mok YK, Yang H, Chen ES (2018) Regulation of transcriptional silencing and chromodomain protein localization at centromeric heterochromatin by histone H3 tyrosine 41 phosphorylation in fission yeast. Nucleic Acids Res 46:189–202. https://doi.org/10.1093/nar/gkx1010
Robellet X, Vanoosthuyse V, Bernard P (2017) The loading of condensin in the context of chromatin. Curr Genet 63:577–589. https://doi.org/10.1007/s00294-016-0669-0
Sadaie M, Iida T, Urano T, Nakayama J (2004) A chromodomain protein, Chp1, is required for the establishment of heterochromatin in fission yeast. EMBO J 23:3825–3835. https://doi.org/10.1038/sj.emboj.7600401
Sadaie M, Kawaguchi R, Ohtani Y, Arisaka F, Tanaka K, Shirahige K, Nakayama J (2008) Balance between distinct HP1 family proteins controls heterochromatin assembly in fission yeast. Mol Cell Biol 28:6973–6988
Salzberg AC, Harris-Becker A, Popova EY, Keasey N, Loughran TP, Claxton DF, Grigoryev SA (2017) Genome-wide mapping of histone H3K9me2 in acute myeloid leukemia reveals large chromosomal domains associated with massive gene silencing and sites of genome instability. PLoS One 12:e0173723. https://doi.org/10.1371/journal.pone.0173723
Seo HD, Kwon CS, Lee D (2018) The 19S proteasome regulates subtelomere silencing and facultative heterochromatin formation in fission yeast. Curr Genet 64:741–752. https://doi.org/10.1007/s00294-017-0792-6
Shilatifard A (2006) Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem 75:243–269
Simon JA, Kingston RE (2009) Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 10:697–708. https://doi.org/10.1038/nrm2763
Sugiyama T, Cam H, Verdel A, Moazed D, Grewal SI (2005) RNA-dependent RNA polymerase is an essential component of a self-enforcing loop coupling heterochromatin assembly to siRNA production. Proc Natl Acad Sci USA 102:152–157
Sugiyama T, Cam HP, Sugiyama R, Noma K, Zofall M, Kobayashi R, Grewal SI (2007) SHREC, an effector complex for heterochromatic transcriptional silencing. Cell 128:491–504. https://doi.org/10.1016/j.cell.2006.12.035
Suzuki S, Kato H, Suzuki Y, Chikashige Y, Hiraoka Y, Kimura H, Nagao K, Obuse C, Takahata S, Murakami Y (2016) Histone H3K36 trimethylation is essential for multiple silencing mechanisms in fission yeast. Nucleic Acids Res 44:4147–4162. https://doi.org/10.1093/nar/gkw008
Thon G, Verhein-Hansen J (2000) Four chromo-domain proteins of Schizosaccharomyces pombe differentially repress transcription at various chromosomal locations. Genetics 155:551–568
Ting DT et al (2011) Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 331:593–596. https://doi.org/10.1126/science.1200801
Trojer P, Reinberg D (2007) Facultative heterochromatin: is there a distinctive molecular signature? Mol Cell 28:1–13
Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, Moazed D (2004) RNAi-mediated targeting of heterochromatin by the RITS complex. Science 303:672–676
Verdel A, Moazed D (2005) RNAi-directed assembly of heterochromatin in fission yeast. FEBS Lett 579:5872–5878. https://doi.org/10.1016/j.febslet.2005.08.083
Xhemalce B, Kouzarides T (2010) A chromodomain switch mediated by histone H3 lys 4 acetylation regulates heterochromatin assembly. Genes Dev 24:647–652. https://doi.org/10.1101/gad.1881710
Yang J, Li F (2017) Are all repeats created equal? Understanding DNA repeats at an individual level. Curr Genet 63:57–63. https://doi.org/10.1007/s00294-016-0619-x
Yu R, Wang X, Moazed D (2018) Epigenetic inheritance mediated by coupling of RNAi and histone H3K9 methylation. Nature 558:615–619. https://doi.org/10.1038/s41586-018-0239-3
Zhang K, Mosch K, Fischle W, Grewal SI (2008) Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat Struct Mol Biol 15:381–388. https://doi.org/10.1038/nsmb.1406
Acknowledgements
We thank members of the Chen Lab for discussion; Rebecca Jackson and Hugh P. Cam for editing the manuscript. This work was supported by a Singapore Ministry of Education Tier 1 Grant (R-183-000-389-112).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by M. Kupiec.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ren, B., Chen, E.S. Regulation of centromeric heterochromatin in the cell cycle by phosphorylation of histone H3 tyrosine 41. Curr Genet 65, 829–836 (2019). https://doi.org/10.1007/s00294-019-00962-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-019-00962-2