
Abstract
Recent advances on antibacterial activity of peptidoglycan recognition proteins (PGRPs) offer some insight into how innate immunity has retained its antimicrobial effectiveness for millions of years with no frequent emergence of resistant strains. First, PGRP can bind to multiple components of bacterial envelope (peptidoglycan, lipoteichoic acid, and lipopolysaccharide). Second, PGRP simultaneously induces oxidative, thiol, and metal stress responses in bacteria, which individually are bacteriostatic, but in combination are bactericidal. Third, PGRP induces oxidative, thiol, and metal stress responses in bacteria through three independent pathways. Fourth, antibacterial effects of PGRP are enhanced by other innate immune responses. Thus, emergence of PGRP resistance is prevented by bacteriostatic effect and independence of each PGRP-induced stress response, as PGRP resistance would require simultaneous acquisition of three separate mechanisms disabling the induction of all three stress responses. By contrast, each antibiotic has one primary target and one primary antibacterial mechanism, and for this reason resistance to antibiotics can be generated by inhibition of this primary mechanism. Manipulating bacterial metabolic responses can enhance bacterial killing by antibiotics and elimination of antibiotic-tolerant bacteria, but such manipulations do not overcome genetically encoded antibiotic resistance. Pathogens cause infections by evading, inhibiting, or subverting host immune responses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Modern medicine, despite its extensive armamentarium of powerful antibiotics, faces a crisis of ever-growing number of antibiotic-resistant bacteria. Introduction of every new antibiotic is followed by quick emergence of resistant strains. By contrast, innate immunity, which offers germline-encoded immediate protection for the host from infections, has retained its antimicrobial effectiveness for millions of years with no frequent emergence of resistant strains. In fact, innate immunity is the only protection from infections in all invertebrates and plants, and it is still an essential component of immunity in vertebrates. Why is innate immunity less prone to microbial resistance than antibiotics, since they both target conserved essential prokaryotic components not found in eukaryotes? We will address this question using human peptidoglycan recognition proteins (PGRPs) as a model of antibacterial innate immunity proteins.
PGRP targets multiple conserved structures in bacteria
Humans and other mammals have four PGRP proteins, coded by PGLYRP1–4 genes. Mammalian PGRPs are all soluble secreted proteins with both recognition and effector functions (Dziarski et al. 2016b; Royet and Dziarski 2007). PGLYRP1, PGLYRP3, and PGLYRP4 are directly bactericidal for both Gram-positive and Gram-negative bacteria (Lu et al. 2006; Tydell et al. 2002, 2006; Wang et al. 2007), and PGLYRP2 is an enzyme, peptidoglycan amidohydrolase (Gelius et al. 2003; Wang et al. 2003).
Each PGRP has one or two PGRP domains with a binding site specific for muramyl-peptide fragment of bacterial peptidoglycan (Dziarski et al. 2016b; Royet and Dziarski 2007). In Gram-positive bacteria, PGRP preferentially binds to muramyl peptides exposed by peptidoglycan-lytic endopeptidases at the separation sites of the newly formed daughter cells (Kashyap et al. 2011). But in addition, mammalian PGRPs have other binding sites specific for bacterial lipoteichoic acid and lipopolysaccharide located outside the peptidoglycan-binding groove (Sharma et al. 2011; Tydell et al. 2006). Thus, PGRPs also bind uniformly to the entire outer membrane of Gram-negative bacteria (Kashyap et al. 2011).
Because each PGRP is so versatile and can bind multiple bacterial components, bacteria cannot easily change these multiple structures to avoid PGRP binding. This binding of PGRPs to bacteria induces exaggerated stress responses in bacteria and initiates a cascade of events that eventually results in bacterial killing (Kashyap et al. 2011, 2014, 2017).
PGRP induces multiple synergistic antibacterial stress responses
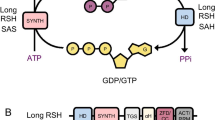
PGRP kills bacteria by simultaneously inducing oxidative, thiol, and metal stress responses in bacteria (Fig. 1). PGRP-induced oxidative stress stems from increased production of hydrogen peroxide (H2O2) and hydroxyl radicals (HO·) (Kashyap et al. 2011, 2014, 2017). PGRP-induced thiol (disulfide) stress stems from depletion of over 90% of intracellular thiols, and metal stress stems from increases in intracellular free Zn2+ and Cu+ (Kashyap et al. 2014, 2017). Induction of all three stress responses is required for PGRP-induced killing. Each stress response individually is only bacteriostatic, and only combined induction of all three stress responses is bactericidal (Kashyap et al. 2014).

PGRP induces oxidative, thiol, and metal stress through independent pathways, which individually are bacteriostatic and together become bactericidal. PGRP induces oxidative stress through a block in respiratory chain, which diverts electrons from respiratory chain NADH oxidoreductases to O2 and generates H2O2. Production of H2O2 depends on increased supply of NADH from glycolysis and tricarboxylic acid (TCA) cycle. PGRP also induces thiol stress (depletion of thiols) and metal stress (increase in intracellular free Zn2+ through influx of extracellular Zn2+), which are mostly independent of oxidative stress and of each other (Kashyap et al. 2014, 2017)
PGRP induces oxidative, thiol, and metal stress through independent pathways
Resistance to PGRPs could easily arise if all three stress responses (i.e., oxidative, thiol, and metal stress) were induced by PGRP through a single pathway. However, PGRP induces oxidative, thiol, and metal stress through three mostly independent pathways (Kashyap et al. 2017). PGRP induces oxidative stress through an increase in central carbon catabolism and simultaneous block in electron transport through the respiratory chain, which results in premature diversion of electrons to oxygen and increased production of H2O2 (Fig. 1). PGRP-induced thiol stress is mostly independent of oxidative stress and metal stress, and may depend on the generation of endogenous electrophiles through a so far unidentified pathway. PGRP-induced metal stress is also independent of oxidative and thiol stress and depends on the increased influx of metals into bacterial cells (Fig. 1) (Kashyap et al. 2017).
Toxicity of oxidative stress is due to excessive production of superoxide anion (O2 −) and H2O2, which oxidize and inactivate solvent-exposed [4Fe–4S] enzyme clusters and inactivate mononuclear iron enzymes by oxidizing Fe-coordinating cysteines or by replacing Fe2+ with Zn2+ (Anjem and Imlay 2012; Gu and Imlay 2013; Jang and Imlay 2007, 2010). H2O2 also reacts with Fe2+ and generates highly toxic HO·, which irreversibly damages DNA, proteins, and other organic molecules (Imlay 2013; Park et al. 2005). Toxicity of thiol stress is due to oxidation of thiols and loss of the redox balance and greater sensitivity to oxidative damage and metal toxicity (Chillappagari et al. 2010; Harrison et al. 2009; Leichert et al. 2003). Toxicity of metal stress depends on the metal involved. Zn and Cu toxicity is in part due to inactivation of solvent-exposed Fe–S clusters (Macomber and Imlay 2009; Xu and Imlay 2012). Zn also inactivates enzymes by replacing Fe2+ in their active sites (Chandrangsu and Helmann 2016; Gu and Imlay 2013), whereas Cu also increases thiol oxidation and sulfhydryl depletion, which magnify thiol stress and protein damage (Chillappagari et al. 2010; Harrison et al. 2009; Macomber and Imlay 2009).
Emergence of PGRP resistance is prevented by bacteriostatic effect and independence of each stress
Although disabling one of the PGRP-induced stress pathways greatly decreases or abolishes PGRP killing, PGRP still remains bacteriostatic. For this reason, full resistance to PGRP does not easily develop, because it would require simultaneous disabling of all three PGRP-induced independent stress responses, which is a very low probability event. Consistent with this notion, mutants fully resistant to antibacterial effects of PGRPs could not be found or generated, despite many efforts (Kashyap et al. 2017).
The initial events that follow PGRP binding to the cell wall in Gram-positive bacteria or to the outer membrane in Gram-negative bacteria and lead to the induction of oxidative, thiol, and metal stress are still not clear. Confocal microscopy indicates that PGRPs do not enter the cytoplasm and induce their lethal effects from their extracellular binding sites (Kashyap et al. 2011). It is not known whether PGRPs stay bound to the cell wall or to the outer membrane, or whether PGRPs also interact with the cytoplasmic membrane, because the resolution of these confocal experiments was not sufficient to make this distinction. However, PGRPs do not permeabilize the cytoplasmic membrane and do not induce osmotic lysis of bacteria (Kashyap et al. 2011; Lu et al. 2006; Wang et al. 2007). But independence of PGRP-induced oxidative, thiol, and metal stress of each other suggests that the initial PGRP-induced events that trigger these stress responses may also be different for each stress response and independent of each other.
Antibiotics and PGRPs have different mechanisms of action
The mechanisms of bacterial killing by PGRPs and antibiotics are different and PGRPs kill bacteria resistant to multiple antibiotics (Kashyap et al. 2014). Whereas PGRPs kill bacteria through simultaneous induction of multiple independent stress responses, each antibiotic has one primary target and one primary antibacterial mechanism. For this reason, resistance to antibiotics can be generated by inhibition of this primary mechanism. For example, antibiotics in each of the major groups, i.e., inhibitors of protein, RNA, DNA, or peptidoglycan synthesis, selectively and immediately inhibit the synthesis of only their respective target, whereas treatment with PGRP results in simultaneous inhibition of all biosynthetic reactions (Kashyap et al. 2011).
Much attention has been recently devoted to determining whether classical antibiotics kill bacteria through induction of oxidative stress or metabolic stress. However, the ability of antibiotics to induce oxidative stress in bacteria and the requirement for induction of oxidative stress for bacterial killing by antibiotics are still controversial (Brynildsen et al. 2013; Dwyer et al. 2014, 2015; Ezraty et al. 2013; Imlay 2015; Keren et al. 2013; Kohanski et al. 2007, 2008; Liu and Imlay 2013; Lobritz et al. 2015; Mahoney and Silhavy 2013). Manipulations of bacterial metabolism and enhancement of oxidative stress in bacteria can increase bacterial killing by antibiotics and help to eliminate antibiotic-tolerant bacteria (Belenky et al. 2015; Dwyer et al. 2014; Lobritz et al. 2015; Meylan et al. 2017). Consistent with this notion, bactericidal antibiotics increase the respiration rate in bacteria (Lobritz et al. 2015). By contrast, PGRPs induce a decrease in the respiration rate in bacteria due to a block in respiratory chain (Kashyap et al. 2017). Moreover, many effects of antibiotics on the metabolism and respiration most likely result from bacterial responses to the primary effects of antibiotics and happen late in antibiotic-induced killing (30–90 min) (Belenky et al. 2015; Kohanski et al. 2007). For this reason, metabolic manipulations do not overcome genetically encoded antibiotic resistance. By contrast, the kinetics of PGRP-induced changes in metabolism and respiration are much faster (5 min) and likely reflect the primary effects of PGRP on bacteria (Kashyap et al. 2017). There are also several additional differences between the mechanisms of bacterial killing by PGRPs and antibiotics (Kashyap et al. 2011, 2014, 2017).
PGRPs are bactericidal only under specific conditions
Natural antibiotics are produced by fungi or bacteria in the soil and have to be active in wide-ranging environmental conditions that the producing organisms cannot control. Therefore, antibacterial effects of antibiotics are generally independent of culture conditions, as long as bacteria can maintain vigorous growth, which is usually required for killing by bactericidal antibiotics. By contrast, PGRPs are produced in the host in very specific cells or tissues. PGLYRP1 is present in neutrophil, eosinophil, and macrophage granules (Dziarski et al. 2003; Liu et al. 2001; Tydell et al. 2002), PGLYRP2 is mostly present in the serum (Hoijer et al. 1996; Wang et al. 2003) and also in the skin, whereas PGLYRP3 and PGLYRP4 are produced on the skin and mucous membranes (especially in the moth and esophagus), and in sweat, sebum, and saliva (Liu et al. 2001; Lu et al. 2006).
Thus, PGRPs only need to be active under very specific conditions of these niches. These body fluids contain significant amounts of Zn and Cu, and PGRPs are only bactericidal in the media with very specific nutrient and ion composition that mimics body secretions or tissue fluids (Wang et al. 2007). The most important is the presence of Zn2+, which is required for bactericidal activity of PGRPs for both Gram-positive and Gram-negative bacteria (Wang et al. 2007), although for killing of Gram-positive bacteria (but not Gram-negative bacteria) Zn2+ can be substituted to some extent by other divalent cations, e.g., Ca2+ (Lu et al. 2006; Wang et al. 2007). PGRP killing also requires a precise type and amount of metabolic activity (Kashyap et al. 2017). For these reasons, PGRPs are usually not bactericidal, but can be bacteriostatic, in common laboratory media or buffers (Liu et al. 2000).
Antibacterial effects of PGRP are enhanced by other innate immune responses
The antibacterial effects of PGRPs are also enhanced by other innate immune responses of the host. For example, phagocytic cells, upon phagocytosis of bacteria, in addition to oxidative killing, pump Cu and Zn into phagolysosomes to enhance bacterial killing (Chandrangsu et al. 2017; German et al. 2013; Palmer and Skaar 2016). As already mentioned, PGRP-induced metal stress depends on the import of these extracellular cations (Kashyap et al. 2017). In response to PGRPs, bacteria up-regulate expression of Cu and Zn exporters (Kashyap et al. 2014). However, PGRPs defeat this bacterial defense by shutting down bacterial respiration and metabolism and depolarizing bacterial membranes (Kashyap et al. 2011, 2017), and thus depriving bacteria of energy and proton motive force needed to drive bacterial Cu and Zn efflux. PGRPs also kill bacteria synergistically with antimicrobial peptides, which are abundant in phagocytic granules and body secretions (Cho et al. 2005; Wang et al. 2007), where PGRPs are present. These synergistic effects with other host defenses further enhance antibacterial effectiveness of PGRPs and prevent development of resistance.
Pathogens evade innate immunity
If bacteria, including pathogens, do not easily develop resistance to PGRPs and other antibacterial innate immunity mechanisms, how do pathogens cause infections? Pathogens are successful in causing infections, because they developed an amazing variety of virulence factors that allow them to evade, inhibit, or otherwise subvert host immune responses (DiRita 2013; Olivos-García et al. 2016; Palmer and Skaar 2016; Reddick and Alto 2014). Greater sensitivity of Pglyrp1-deficient mice to infections with non-pathogenic, but not with pathogenic bacteria (which are both sensitive to PGRP killing in vitro) (Dziarski et al. 2003), suggests that in vivo pathogens can avoid antibacterial effects of PGRP. The ability of PGRPs to control the composition of microbiome also suggests differential antibacterial effects of PGRPs in vivo and differential in vivo sensitivity of various bacteria to PGRPs (Dziarski et al. 2016a, b; Royet et al. 2011). However, how pathogens evade PGRPs in vivo and how PGRPs differentially affect the composition of microbiome are not known and should be a fertile area for future studies.
References
Anjem A, Imlay JA (2012) Mononuclear iron enzymes are primary targets of hydrogen peroxide stress. J Biol Chem 287:15544–15556. doi:10.1074/jbc.M111.330365
Belenky P, Ye JD, Porter CB, Cohen N, Lobritz MA, Ferrante T et al (2015) Bactericidal antibiotics induce toxic metabolic perturbations that lead to cellular damage. Cell Rep 13:968–980. doi:10.1016/j.celrep.2015.09.059
Brynildsen MP, Winkler JA, Spina CS, MacDonald IC, Collins JJ (2013) Potentiating antibacterial activity by predictably enhancing endogenous microbial ROS production. Nat Biotechnol 31:160–165. doi:10.1038/nbt.2458
Chandrangsu P, Helmann JD (2016) Intracellular Zn(II) intoxication leads to dysregulation of the PerR regulon resulting in heme toxicity in Bacillus subtilis. PLoS Genet 12(12):e1006515. doi:10.1371/journal.pgen.1006515
Chandrangsu P, Rensing C, Helmann JD (2017) Metal homeostasis and resistance in bacteria. Nat Rev Microbiol 15:338–350. doi:10.1038/nrmicro.2017.15
Chillappagari S, Seubert A, Trip H, Kuipers OP, Marahiel MA, Miethke M (2010) Copper stress affects iron homeostasis by destabilizing iron-sulfur cluster formation in Bacillus subtilis. J Bacteriol 192:2512–2524. doi:10.1128/JB.00058-10
Cho JH, Fraser IP, Fukase K, Kusumoto S, Fujimoto Y, Stahl GL, Ezekowitz RA (2005) Human peptidoglycan recognition protein S is an effector of neutrophil-mediated innate immunity. Blood 106:2551–2558. doi:10.1182/blood-2005-02-0530
DiRita VJ (2013). The parasite’s way of life. In: Engelberg NC, DiRita V, Dermody TS (eds) Schaechter’s mechanisms of microbial disease, 5th edn, Wolters Kluwer/Lippincott, Williams & Wilkins, Baltimore/Philadelphia, ch 8, pp 117–126
Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N et al (2014) Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci USA 111:E2100–E2109. doi:10.1073/pnas.1401876111
Dwyer DJ, Collins JJ, Walker GC (2015) Unraveling the physiological complexities of antibiotic lethality. Annu Rev Pharmacol Toxicol 55:313–332. doi:10.1146/annurev-pharmtox-010814-124712
Dziarski R, Platt KA, Gelius E, Steiner H, Gupta D (2003) Defect in neutrophil killing and increased susceptibility to infection with non-pathogenic Gram-positive bacteria in peptidoglycan recognition protein-S (PGRP-S)-deficient mice. Blood 102:689–697. doi:10.1182/blood-2002-12-3853
Dziarski R, Park SY, Kashyap DR, Dowd SE, Gupta D (2016a) Pglyrp-regulated gut microflora Prevotella falsenii, Parabacteroides distasonis and Bacteroides eggerthii enhance and Alistipes finegoldii attenuates colitis in mice. PLoS One 11:e0146162. doi:10.1371/journal.pone.0146162
Dziarski R, Royet J, Gupta D (2016b) Peptidoglycan recognition proteins and lysozyme. In: Ratcliffe MJH (ed) Encyclopedia of immunobiology, vol 2. Elsevier, Academic Press, Oxford, pp 389–403
Ezraty B, Vergnes A, Banzhaf M, Duverger Y, Huguenot A, Brochado AR et al (2013) Fe-S cluster biosynthesis controls uptake of aminoglycosides in a ROS-less death pathway. Science 340:1583–1587. doi:10.1126/science.1238328
Gelius E, Persson C, Karlsson J, Steiner H (2003) A mammalian peptidoglycan recognition protein with N-acetylmuramoyl-l-alanine amidase activity. Biochem Biophys Res Commun 306:988–994
German N, Doyscher D, Rensing C (2013) Bacterial killing in macrophages and amoeba: do they all use a brass dagger? Future Microbiol 8:1257–1264. doi:10.2217/fmb.13.100
Gu M, Imlay JA (2013) Superoxide poisons mononuclear iron enzymes by causing mismetallation. Mol Microbiol 89:123–134. doi:10.1111/mmi.12263
Harrison JJ, Tremaroli V, Stan MA, Chan CS, Vacchi-Suzzi C, Heyne BJ et al (2009) Chromosomal antioxidant genes have metal ion-specific roles as determinants of bacterial metal tolerance. Environ Microbiol 11:2491–2509. doi:10.1111/j.1462-2920.2009.01973.x
Hoijer MA, Melief MJ, Keck W, Hazenberg MP (1996) Purification and characterization of N-acetylmuramoyl-l-alanine amidase from human plasma using monoclonal antibodies. Biochim Biophys Acta 1289:57–64
Imlay JA (2013) The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11:443–454. doi:10.1038/nrmicro3032
Imlay JA (2015) Diagnosing oxidative stress in bacteria: not as easy as you might think. Curr Opin Microbiol 24:124–131. doi:10.1016/j.mib.2015.01.004
Jang S, Imlay JA (2007) Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J Biol Chem 282:929–937. doi:10.1074/jbc.M607646200
Jang S, Imlay JA (2010) Hydrogen peroxide inactivates the Escherichia coli Isc iron-sulphur assembly system, and OxyR induces the Suf system to compensate. Mol Microbiol 78:1448–1467. doi:10.1111/j.1365-2958.2010.07418.x
Kashyap DR, Wang M, Liu L-H, Boons G-J, Gupta D, Dziarski R (2011) Peptidoglycan recognition proteins kill bacteria by activating protein-sensing two-component systems. Nat Med 17:676–683. doi:10.1111/j.1365-2958.2010.07418.x
Kashyap DR, Rompca A, Gaballa A, Helmann JD, Chan J, Chang CJ et al (2014) Peptidoglycan recognition proteins kill bacteria by inducing oxidative, thiol, and metal stress. PLoS Pathog 10:e1004280. doi:10.1371/journal.ppat.1004280
Kashyap DR, Kuzma M, Kowalczyk DA, Gupta D, Dziarski R (2017) Bactericidal peptidoglycan recognition protein induces oxidative stress in Escherichia coli through a block in respiratory chain and increase in central carbon catabolism. Mol Microbiol. doi:10.1111/mmi.13733
Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K (2013) Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339:1213–1216. doi:10.1126/science.1232688
Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ (2007) A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810. doi:10.1016/j.cell.2007.06.049
Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ (2008) Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135:679–690. doi:10.1016/j.cell.2008.09.038
Leichert LIO, Scharf C, Hecker M (2003) Global characterization of disulfide stress in Bacillus subtilis. J Bacteriol 185:1967–1975
Liu Y, Imlay JA (2013) Cell death from antibiotics without the involvement of reactive oxygen species. Science 339:1210–1213. doi:10.1126/science.1232751
Liu C, Gelius E, Liu G, Steiner H, Dziarski R (2000) Mammalian peptidoglycan recognition protein binds peptidoglycan with high affinity, is expressed in neutrophils and inhibits bacterial growth. J Biol Chem 275:24490–24499. doi:10.1074/jbc.M001239200
Liu C, Xu Z, Gupta D, Dziarski R (2001) Peptidoglycan recognition proteins: a novel family of four human innate immunity pattern recognition molecules. J Biol Chem 276:34686–34694. doi:10.1074/jbc.M105566200
Lobritz MA, Belenky P, Porter CB, Gutierrez A, Yang JH, Schwarz EG et al (2015) Antibiotic efficacy is linked to bacterial cellular respiration. Proc Natl Acad Sci USA 112:8173–8180. doi:10.1073/pnas.1509743112
Lu X, Wang M, Qi J, Wang H, Li X, Gupta D, Dziarski R (2006) Peptidoglycan recognition proteins are a new class of human bactericidal proteins. J Biol Chem 281:5895–5907. doi:10.1074/jbc.M511631200
Macomber L, Imlay JA (2009) The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc Natl Acad Sci USA 106:8344–8349. doi:10.1073/pnas.0812808106
Mahoney TF, Silhavy TJ (2013) The Cpx stress response confers resistance to some, but not all, bactericidal antibiotics. J Bacteriol 195:1869–1874. doi:10.1128/JB.02197-12
Meylan S, Porter CB, Yang JH, Belenky P, Gutierrez A, Lobritz MA et al (2017) Carbon sources tune antibiotic susceptibility in Pseudomonas aeruginosa via tricarboxylic acid cycle control. Cell Chem Biol 24:195–206. doi:10.1016/j.chembiol.2016.12.015
Olivos-García A, Saavedra E, Nequiz M, Santos F, Luis-García ER, Gudiño M, Pérez-Tamayo R (2016) The oxygen reduction pathway and heat shock stress response are both required for Entamoeba histolytica pathogenicity. Curr Genet 62:295–300. doi:10.1007/s00294-015-0543-5
Palmer LD, Skaar EP (2016) Transition metals and virulence in bacteria. Annu Rev Genet 50:67–91. doi:10.1146/annurev-genet-120215-035146
Park S, You X, Imlay JA (2005) Substantial DNA damage from submicromolar intracellular hydrogen peroxide detected in Hpx− mutants of Escherichia coli. Proc Natl Acad Sci USA 102:9317–9322. doi:10.1073/pnas.0502051102
Reddick LE, Alto NM (2014) Bacteria fighting back: how pathogens target and subvert the host innate immune system. Mol Cell 54:321–328. doi:10.1016/j.molcel.2014.03.010
Royet J, Dziarski R (2007) Peptidoglycan recognition proteins: pleiotropic sensors and effectors of antimicrobial defences. Nat Rev Microbiol 5:264–277. doi:10.1038/nrmicro1620
Royet J, Gupta D, Dziarski R (2011) Peptidoglycan recognition proteins: modulators of the microbiome and inflammation. Nat Rev Immunol 11:837–851. doi:10.1038/nri3089
Sharma P, Dube D, Singh A, Mishra B, Singh N, Sinha M et al (2011) Structural basis of recognition of pathogen-associated molecular patterns and inhibition of proinflammatory cytokines by camel peptidoglycan recognition protein. J Biol Chem 286:16208–16217. doi:10.1074/jbc.M111.228163
Tydell CC, Yount N, Tran D, Yuan J, Selsted ME (2002) Isolation, characterization, and antimicrobial properties of bovine oligosaccharide-binding protein. A microbicidal granule protein of eosinophils and neutrophils. J Biol Chem 277:19658–19664. doi:10.1074/jbc.M200659200
Tydell CC, Yuan J, Tran P, Selsted ME (2006) Bovine peptidoglycan recognition protein-S: antimicrobial activity, localization, secretion and binding properties. J Immunol 176:1154–1162
Wang Z-M, Li X, Cocklin RR, Wang M, Wang M, Fukase K et al (2003) Human peptidoglycan recognition protein-L is an N-acetylmuramoyl-l-alanine amidase. J Biol Chem 278:49044–49052. doi:10.1074/jbc.M307758200
Wang M, Liu LH, Wang S, Li X, Lu X, Gupta D, Dziarski R (2007) Human peptidoglycan recognition proteins require zinc to kill both Gram-positive and Gram-negative bacteria and are synergistic with antibacterial peptides. J Immunol 178:3116–3125
Xu FF, Imlay JA (2012) Silver(I), mercury(II), cadmium(II), and zinc(II) target exposed enzymic iron-sulfur clusters when they toxify Escherichia coli. Appl Environ Microbiol 78:3614–3621. doi:10.1128/AEM.07368-11
Acknowledgements
This work was supported by a Grant (AI120962) from the United States Public Health Service National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kupiec.
Rights and permissions
About this article

Cite this article
Dziarski, R., Gupta, D. How innate immunity proteins kill bacteria and why they are not prone to resistance. Curr Genet 64, 125–129 (2018). https://doi.org/10.1007/s00294-017-0737-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-017-0737-0