
Abstract
The main objectives of this study were to evaluate genetic composition of Geosmithia morbida populations in the native range of black walnut and provide a better understanding regarding demography of the pathogen. The fungus G. morbida, and the walnut twig beetle, Pityophthorus juglandis, have been associated with a disease complex of black walnut (Juglans nigra) known as thousand cankers disease (TCD). The disease is manifested as branch dieback and canopy loss, eventually resulting in tree death. In 2010, the disease was detected in black walnut in Tennessee, and subsequently in Virginia and Pennsylvania in 2011 and North Carolina in 2012. These were the first incidences of TCD east of Colorado, where the disease has been established for more than a decade on indigenous walnut species. A genetic diversity and population structure study of 62 G. morbida isolates from Tennessee, Pennsylvania, North Carolina and Oregon was completed using 15 polymorphic microsatellite loci. The results revealed high haploid genetic diversity among seven G. morbida populations with evidence of gene flow, and significant differentiation among two identified genetic clusters. There was a significant correlation between geographic and genetic distance. Understanding the genetic composition and demography of G. morbida can provide valuable insight into recognizing factors affecting the persistence and spread of an invasive pathogen, disease progression, and future infestation predictions. Overall, these data support the hypotheses of two separate, highly diverse pathogen introductions into the native range of black walnut.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the past decade, black walnut (Juglans nigra) trees throughout western North America have exhibited widespread branch dieback and canopy loss, causing substantial tree mortality (Kolarik et al. 2011). The fungus, Geosmithia morbida (Ascomycota:Hypocreales:Bionectriaceae), vectored by the walnut twig beetle, Pityophthorus juglandis Blackman (Coleoptera:Curculionidae:Scolytinae), has been associated with this devastating disease outbreak known as thousand cankers disease (TCD) (Kolarik et al. 2011; Tisserat et al. 2009). Although P. juglandis is broadly distributed in the US, ranging from Idaho to Arizona and from Oregon to Pennsylvania (Seybold et al. 2012), the origin of G. morbida is still ambiguous. G. morbida is the first species of its genus to be considered a plant pathogen (Kolarik et al. 2011). In 2010, the disease was detected in the native range of black walnut including Tennessee (Grant et al. 2011) and subsequently in Virginia (Hansen et al. 2011) and Pennsylvania in 2011 (Penn State-Penn State Cooperative Extension 2011) and North Carolina (Great Smoky Mountains National Park) in 2012 (Hadziabdic et al. 2013). As a tree native to eastern and midwestern regions of the United States, black walnut is important both ecologically as a food source for forest animals, and economically as a specialty crop used for nuts and timber (Grant et al. 2011). In order to provide a better understanding regarding demography of this devastating disease, it is essential to evaluate genetic composition of G. morbida populations in the native range of black walnut, which was the main objective of this research.
Infected trees initially display wilting and yellowing foliage (flagging), branch dieback and eventual die-off within 3–4 years after symptoms are detected (Kolarik et al. 2011; Tisserat et al. 2009). Rapid tree mortality is the result of movement and feeding by P. juglandis in the galleries in the phloem, followed by pathogen canker development near those galleries (Kolarik et al. 2011; Tisserat et al. 2009). As the beetles develop tunnels underneath the black walnut bark, and spread throughout the tree by creating galleries, multiple dark brown to black cankers form that coalesce and girdle twigs and branches. Canker development surrounds the galleries in the phloem as a result of colonization by G. morbida. Formation of numerous, assumed to be annual cankers led to naming this pathogen–insect complex thousand cankers disease (Cranshaw and Tisserat 2008). Fungal colonization ultimately destroys the secondary cambium and phloem resulting in tree mortality. All walnut species examined (J. ailantifolia, J. californica, J. cinerea, J.hindsii, J. major, J. mandshurica, J. microcarpa, J. nigra, and J. regia) were susceptible to TCD after inoculation with G. morbida (Utley et al. 2013). However, J. nigra inoculated with G. morbida resulted in the most severe canker development, presumably making it the most susceptible of the walnut species.
Despite the devastating impact caused by TCD complex on black walnuts, including massive mortality and possible extinction of large populations, information on the genetic composition of this haploid fungal pathogen is limited, and very little is known about the epidemiology and impact of this disease on walnut trees. Presently, the only molecular study of G. morbida populations was conducted in the western US, where black walnuts are planted but not native. Eight rDNA ITS haplotypes were detected using 37 G. morbida isolates from different species of walnuts including J. nigra, J. major, J. californica and J. hindsii (Kolarik et al. 2011). G. morbida isolates from the western US are genetically diverse, but constitute a single morphologically and ecologically homogenous species, which is significantly different from other described Geosmithia species (Kolarik et al. 2011). However, no large-scale survey regarding G. morbida populations in the native range of black walnuts and disease spread has been described.
Currently, the estimated value of standing black walnut trees in the US is $568 billion (USDA-APHIS 2009; USDA-FS-PPQ 2012). Black walnut is a valuable nut and timber species in the East, and extensive mortality of these populations would have a negative impact on biodiversity and conservation efforts of this native North American tree species. At this time, disease management is unavailable and no pesticides or control methods exist to prevent the spread of TCD. If this devastating disease complex becomes established throughout the native region of black walnut, the economic and environmental consequences could be enormous. Although some progress has been made in understanding the life cycle of P. juglandis, details regarding spatial distribution, genetic diversity and biology of the pathogen–insect complex associated with TCD in the native range of black walnut are limited. We hypothesize that the latest detection of G. morbida in Tennessee, Virginia, Pennsylvania and North Carolina are recent, separate introductions, and that the disease complex has existed undetected for a number of years. However, how this movement from west to east occurred has not been explained. Revealing the genetic structure of the pathogen populations can provide an insight into their evolutionary potential and explain which pathogen groups can provide the highest risk and largest mortality rates (McDonald and Linde 2002; Stefansson et al. 2012). Large-scale mortality of native species and severe die-offs often result in alteration of forest communities, disturbance of ecosystem processes and rapid decline of genetic variation (Hall et al. 2002; Becker et al. 2008; Elliott and Swank 2008; Eschtruth and Battles 2008). Decline of genetic diversity has potentially undesirable effects on the ability of tree populations to regenerate and respond to future changes in disease selection pressures. Therefore, we hypothesize that that existing populations of the pathogen are diverse and capable of causing significant suppression and disturbance that can contribute to local and regional declines of native black walnut trees. In this study, the objective was to evaluate genetic composition of G. morbida populations in the native range of black walnut and provide a better understanding regarding this devastating disease.
Materials and methods
G. morbida isolates
Sixty-two G. morbida isolates were collected from infested areas located in Tennessee, Pennsylvania, North Carolina (Great Smoky Mountains National Park-GRSM) and Oregon (7 subpopulations—Anderson, Blount, Knox, Loudon, Sevier, GRSM and Oregon) (Table 1). To allow analysis of the population structure, a single Pennsylvania isolate was pooled with the Anderson group, which consisted of two isolates. All samples were collected from May to October of 2010, 2011, and 2012, which is the optimum timing for both beetle and pathogen life cycles in the eastern United States based on our preliminary data (not shown). Branches from symptomatic trees (5–10 cm in diameter and 15–30 cm long) were collected, placed in plastic bags and labeled with the following information: county, state, sample location with GPS coordinates, date, and collector’s name. Labeled samples were placed in an environmental chamber (12 °C) and processed, usually within 1–3 days, in the laboratory.
The outer bark of symptomatic branches was removed to expose cankers and/or P. juglandis. Small bark chips (5–10 mm long and 3–5 mm wide from canker margins) were cultured on 1/10 strength potato dextrose agar medium amended with 30 mg/l streptomycin sulfate and 30 mg/l chlorotetracycline HCl (1/10 PDA +) in 100-mm petri dishes. Dishes were incubated on a 12 h dark/light cycle at 22 °C for 6–10 days. Besides canker samples, additional cultures were obtained by direct plating (streaking beetle parts) of infested P. juglandis and P. juglandis frass. A third group of cultures was obtained by washing P. juglandis in 100 μl of sterile water in 2 ml microcentrifuge tubes and vortexed for 15 s. A pipet was then used to distribute the water suspension in equal amounts onto 1/10 PDA + medium contained in 100 mm petri dishes. The dishes were wrapped with parafilm and incubated as described above for canker samples. Single-spore G. morbida isolates were tentatively identified as G. morbida using culture morphology, and conidiophores and conidia characteristics (Fig. 1).

Geosmithia morbida colonies grown on 1/10 PDA (a). Light micrograph of G. morbida conidia (b) and conidiophores (c). Images b and c courtesy of Dr. A. Windham, University of Tennessee
The identities of morphologically identified G. morbida samples were confirmed using ITS1 (5′-TCCGTAGGTGAACCTGCGG) and ITS4 (5′-TCCTCCGCTTATTGATATGC) regions (White et al. 1990). Polymerase chain reaction (PCR) amplifications were performed in 25 μl reaction mixtures containing 10 μl DNA template (2 ng/μl), 2.5 μl of GeneAmp 10× PCR Buffer II (Applied Biosystems, Branchburg, NJ, USA), 2.5 μl of 25 mM MgCl2, 2.5 μl of each 2 mM of dNTPs, 1.25 μl of 10 μM ITS1, 1.25 μl of 10 μM ITS4, 1.25 μl of 5 % dimethyl sulphoxide (DMSO), 1 μl of 5 U/μl AmpliTaq Gold® DNA polymerase (Applied Biosystems), and 2.75 μl of sterile, nanopure water. Amplification reactions were temperature-cycled in 0.2 ml 8-strip PCR tubes using an Eppendorf Autorisierter Thermocycler (Eppendorf AG, Hamburg, Germany) following an initial denaturation step of 2 min at 94 °C, followed by 40 cycles of 94 °C for 30 s, 57 °C for 1 min, 72 °C for 1.5 min, and a final extension at 72 °C for 7 min. Following amplification, samples were sequenced using ABI 3100, and compared with other G. morbida sequences in GenBank. A voucher specimen for G. morbida has been deposited with the American Type Culture Collection (ATCC MYA-4903). Branch samples infested with P. juglandis were identified by Drs. Jerome F. Grant and Gregory J. Wiggins (University of Tennessee) and voucher samples confirmed by Dr. Whitney Cranshaw (Colorado State University) (Grant et al. 2011).
DNA extraction, PCR amplification and genotyping of G. morbida using microsatellite loci
Fungal DNA was extracted from G. morbida samples grown in liquid cultures (1/10 Potato Dextrose Broth) at room temperature for 5–7 days. Fungal mycelia were ground in liquid nitrogen using an autoclaved mortar and pestle. DNA was extracted using a DNeasy Plant Mini Kit (Qiagen, Valencia, CA) according to a modified version of the manufacturer’s published protocol, which was the addition of 1.5 % polyvinylpyrrolidone (PVP) to the lysis (AP1) buffer. PVP acts a strong antioxidant that prevents DNA binding with polyphenols and therefore increases the quality of extracted fungal DNA by removing undesirable impurities (Maliyakal 1992; Nunes et al. 2011). The concentration and purity of DNA samples were measured with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE, USA) before PCR amplification for microsatellite analyses.
Fifteen polymorphic microsatellite loci from G. morbida (Hadziabdic et al. 2012) were used to evaluate pathogen population structure and genetic diversity (Table 2). PCR amplifications were performed in 10 μl reaction mixtures containing 1 μl DNA template (2 ng/μl), 1 μl of GeneAmp 10× PCR Buffer II (Applied Biosystems, Branchburg, NJ, USA), 1 μl of 25 mM MgCl2, 1 μl of each 2 mM of dNTPs, 1 μl of 2.5 μM forward and reverse specific microsatellite primers, 0.08 μl of 5 U/μl AmpliTaq Gold® DNA polymerase (Applied Biosystems), and 4.92 μl of sterile, nanopure water. Amplification reactions were temperature-cycled in 96-well plates using an Eppendorf Autorisierter Thermocycler (Eppendorf AG, Hamburg, Germany) with the following program: 94 °C for 3 min, followed by 35 cycles of 94 °C for 40 s, 55 °C for 40 s, 72 °C for 30 s, and a final extension at 72 °C for 4 min. PCR products were analyzed with the QIAxcel Capillary Electrophoresis System (Qiagen, Valencia, CA, USA) using an internal 25-bp DNA size marker. With the QIAxcel system, data are automatically recorded and exported using BioCalculator™ software, which provides both a gel view and an electropherogram of the separated PCR products (alleles).
Data analyses
FLEXIBIN (Amos et al. 2007) was used for automated binning of allelic data by converting raw allele lengths into allele classes. The program is computed using a simple algorithm coded in Visual Basic as an Excel Macro. Upon finding the best-fit values (best binning parameters), raw allelic data are replaced with their repeat unit equivalents and a graphical output file with additional summary statistics is generated. Here, binned data were presented and used for further analyses.
Program GenAlEx 6.5 (Peakall and Smouse 2012) was used for computation of standard measures of genetic diversity among the 62 G. morbida isolates. Analyses included the number of different alleles, private allele numbers, Shannon’s information index (I), haploid genetic diversity (h) and unbiased haploid genetic diversity (uh) for all populations across all tested loci. Pairwise population ΦPT (analogous to standardized F ST for haploid data) and haploid number of migrants (N m) values were obtained after 9,999 permutations among seven G. morbida subpopulations. N m values can be interpreted as the effective number of migrants or gene flow exchanged between subpopulations per generation (Wright 1969). Principal coordinate analysis (PCoA) was carried out to visualize structure in the relationships among G. morbida isolates using a covariance haploid matrix with data standardization. This multivariate analysis identifies major patterns among a multivariate dataset and creates two-dimensional scatter plots by using the first and second principal coordinates. Nei’s standard genetic distance (Ds) matrix (Nei 1972) between G. morbida subpopulations was computed using the program POPULATIONS 1.2.32 (Langella 1999). Bootstrapping was carried out using 500 replications and the unweighted pair group method with arithmetic mean (UPGMA) clustering analysis visualized using the TreeView software package (Page 1996).
The population structure across different G. morbida subpopulations was further evaluated using Bayesian cluster analysis (Pritchard et al. 2000), analysis of molecular variance (AMOVA) (Excoffier et al. 1992) and Mantel test for isolation by distance (Smouse et al. 1986). The Bayesian-based clustering method was applied to infer the number of subpopulations among G. morbida isolates using the program STRUCTURE version 2.3.4 (Pritchard and Donnelly 2001; Hubisz et al. 2009). The program groups individuals according to their multilocus genotypes without making assumptions about the relationship between sample sites and population structure. It also attempts to find population groupings that are not in disequilibrium (Pritchard and Donnelly 2001). Evanno’s method (Evanno et al. 2005) was used to identify the appropriate number of clusters using the ad hoc statistic Δk. Ten independent runs for each k value between one and eight were performed at 100,000 MCMC (Markov chain Monte Carlo) repetitions and a burn-in period of 100,000 iterations using an admixture model with correlated allele frequencies (assuming no prior information of population origin).
Program Arlequin version 3.5.1.2 (Excoffier and Lischer 2010) was used to compute the hierarchical distribution of genetic variation of all G. morbida subpopulations using AMOVA (analysis obtained using 99,999 permutations). Genetic structure was analyzed by combining all subpopulations (Anderson, Blount, Knox, Loudon, Sevier, GRSM and Oregon) into one hierarchical group as well as partitioning subpopulations into two groups, based on STRUCTURE analysis. Although AMOVA can complement STRUCTURE findings by providing the percentage of variation for the groups identified by STRUCTURE, these outcomes should not be taken as independent confirmation of implied hierarchical structure.
To evaluate whether or not geographical structure is best explained by hierarchical clustering, correlation between pairwise genetic and geographic distance was estimated using GenAlEx 6.5, and statistical significance was evaluated using Mantel test using 9,999 permutations. Mantel tests were performed following Smouse et al. (1986), which estimates matrix correspondence using a measure (Rxy) analogous to an autocorrelation coefficient.
Since TCD epidemics are a recent introduction into the native region of black walnut, we used program BOTTLENECK 1.2 (Piry et al. 1999) to test for any indication of population bottleneck as an excess or deficit (a recent population bottleneck or expansion, respectively) of genetic diversity, relative to the number of alleles present in G. morbida subpopulations. The program assumes that populations with recent and sudden decline in effective population size generally result in higher gene diversity (equilibrium heterozygosity) than predicted by the number of alleles simply because rare alleles are lost rapidly while gene diversity decreases more slowly. For this analysis, subpopulations were aggregated into regional populations that could be supported by analyses of population structure and computed using infinite allele model (IAM), stepwise mutation model (SMM) and a two-phased model of mutation (TPM) (Cornuet and Luikart 1996; Piry et al. 1999). The significance of gene diversity deficit or excess was evaluated using SIGN and the one-tailed Wilcoxon test with 10,000 replications.
Results
Fifteen polymorphic microsatellite loci used for the genotyping of 62 G. morbida isolates indicated 52 haplotypes and revealed that the total of 115 alleles across all loci ranged from 4 to 17 alleles per locus. Presence of private alleles (an allele found in only one population) was observed across all loci except GS058, and across six out of seven subpopulations—Anderson, Blount, Knox, Loudon, Oregon and GRSM. Oregon and GRSM subpopulations had the highest number of loci with private alleles, ranging from 3 to 6. Haploid genetic diversity across all seven subpopulations was 0.42 (SD ± 0.02), ranging from 0.30 (SD ± 0.07) in the Loudon subpopulation to 0.60 (SD ± 0.04) for the Blount subpopulation (Table 3). The same differences across populations were observed for Shannon’s information index—the highest values were observed across isolates from the Blount subpopulation when compared to the Loudon subpopulation [1.17 (SD ± 0.13) and 0.44 (SD ± 0.10)], respectively (Table 3). The total unbiased haploid diversity across all populations was 0.52 (SD ± 0.03).
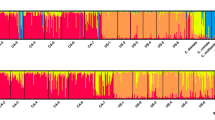
Results obtained using the program STRUCTURE indicated a clear maximum of two different clusters (Δk at k = 2) using Evanno’s method (Evanno et al. 2005) (Fig. 2). Two clusters included GRSM and Oregon isolates as one group, and Anderson, Blount, Knox, Loudon and Sevier (all TN isolates) isolates as the second group (Fig. 2a, b). The multiple runs of the same k were used in our analyses to ensure highly consistent individual assignment probabilities. The genetic distances among seven G. morbida subpopulations were computed using Nei’s standard genetic distance Ds and visualized as UPGMA tree, further supporting population clustering patterns revealed by STRUCTURE (Fig. 2b). Two clusters identified by STRUCTURE corresponded well to geographic regions, which were also in agreement with the PCoA scatter plot (Fig. 3). The first three principal coordinates from the genetic distance matrix explained 23.65, 17.81 and 16.45 % of variation of across the two identified groups, respectively.

Map of Geosmithia morbida sampling locations. Data include 62 isolates from seven different populations—all populations except Oregon (OR) were collected in the native region of black walnut. Pie charts represent the average inferred ancestry of individuals for each cluster identified by STRUCTURE (a). STRUCTURE results revealed two distinct clusters among Geosmithia morbida populations representing five subpopulations from Tennessee (Anderson, Blount, Knox, Loudon and Sevier) as one cluster (orange) and Oregon and North Carolina/Great Smoky Mountains National Park (GRSM) as the second cluster (blue) (a). UPGMA dendogram was constructed using Nei’s genetic distance Ds among seven G. morbida populations (b)

Principal coordinate analysis (PCoA) of Geosmithia morbida populations. Analysis revealed two groups among examined populations, which was supported by the STRUCTURE analysis (Δk = 2). Two different clusters were identified and color coded—a red circle representing Oregon and North Carolina/Great Smoky Mountains National Park (GRSM) as one cluster and a green circle representing five populations from Tennessee (Anderson, Blount, Knox, Loudon and Sevier)
Two different AMOVA analyses were conducted. The first analysis included all subpopulations combined as one hierarchical group and the second AMOVA analysis partitioned subpopulations into two different clusters established by STRUCTURE. Our findings indicated that most of the genetic variation between the seven G. morbida subpopulations can be explained by individual variation rather than divergence across sampling localities, 76.32 and 23.68 %, respectively (P < 0.001) (Table 4). When the genetic variance was partitioned into two distinct clusters established on the basis of the Bayesian and distance analyses, results revealed similar findings. A majority of the differentiation can be explained by variation among individual isolates within populations (58.57 %, P < 0.001). Differences among populations within two clusters accounted for 8.84 % (P < 0.001) of variation among two clusters, and explained 32.59 % of variation (P < 0.047) (Table 4).
A pairwise, individual-based analysis of isolation by distance (IBD) across G. morbida isolates indicated a significant positive relationship between haploid genetic and geographical distance (r = 0.478, P < 0.001) (Fig. 4). When isolates were partitioned into two clusters inferred by STRUCTURE and analyzed separately, subpopulations consisting of TN isolates showed no evidence of IBD among five populations of G. morbida (r = −0.03, P < 0.293). The second cluster (Oregon and GRSM) indicated significant isolation by distance (r = 0.78, P < 0.018), supporting the interpretation of spatial structure within the group (data not shown). P values were obtained using 9,999 randomizations. Geographic distance values are in log scale (Fig. 4).

Scatterplot of pairwise haploid genetic distances vs. geographical distances indicated significant isolation by distance among 62 Geosmithia morbida isolates. Geographic distance values are in log scale
Pairwise comparison of genetic differentiation between the seven subpopulations of G. morbida indicated significant genetic differentiation among G. morbida-identified clusters, therefore further supporting findings obtained by STRUCTURE and AMOVA analyses (Table 5). OR and GRSM groups appeared significantly differentiated from the rest of the populations, resulting in high Φ PT (analogous to standardized F ST for haploid data) values among all subpopulations (0.302–0.586, P < 0.05) with the exception of OR-Anderson isolates, which was not significant (0.407, P > 0.05) (Table 5). Among TN subpopulations, genetic differentiation was comparatively lower, ranging from 0.061 to 0.298 (P < 0.05). For the number of migrants (N m), a similar trend was observed. The highest gene flow was observed between TN subpopulations with the lowest differentiation values (0.046 and 0.047, P > 0.05) (N m = 10.314 and 10.208 between Blount-Anderson and Blount-Sevier, respectively) when compared to OR and GRSM groups (N m = 0.354 for GRSM-Loudon) (Table 5). This finding indicates that TN subpopulations shared more genotypes with higher gene flow among those populations when compared to GRSM and OR subpopulations (Fig. 2a).
Pathogens that undergo severe reduction in population size such as population bottleneck are less diverse and adaptable. Population bottleneck among G. morbida individuals was tested using two previously identified genetic clusters in the program BOTTLENECK. The SIGN and one-tail Wilcoxon’s tests were used to detect any significant excess or deficit in gene diversity (Cornuet and Luikart 1996). In the Oregon-GRSM cluster, one locus (GS058) was monomorphic and allele frequency did not follow a normal L-shaped distribution, indicating that the populations were not at mutation–drift equilibrium. In the TN cluster, there was a significant excess in gene diversity under IAM (P = 0.04) only, indicating a population bottleneck. However, we did not find significant evidence of a recent bottleneck under any other model tested. The two population clusters indicated by STRUCTURE each had 5 and 6 loci with heterozygosity deficiency, and 10 and 8 loci with heterozygosity excess, but they were not significant under sign test and assumptions that all loci fit IAM (P = 0.30, P = 0.52, respectively, for TN and Oregon-GRSM cluster). The lack of significance across all tested models may be due to an insufficient number of loci (<20) (Cornuet and Luikart 1996) as well as limited number of isolates in Oregon-GRSM cluster.
Discussion
Knowledge regarding the genetic structure of pathogen populations can provide an insight into their evolutionary potential (McDonald and Linde 2002) and elucidate host–pathogen interactions, co-evolution and potential decline of host defense mechanisms. The results obtained in this study indicated high levels of genetic diversity and population differentiation between two genetic clusters, with evidence of gene flow between TN subpopulations of G. morbida. Similar findings of high genetic diversity were observed among G. morbida isolates from the western US (Kolarik et al. 2011). Tennessee subpopulations were closely related and shared more genotypes, suggesting a recent common ancestor for those isolates. Vector transmission of fungal pathogens such as G. morbida ensures population continuity from generation to generation, therefore influencing the phylogenetic history of fungal associates (Kolarik et al. 2008). Bayesian clustering and distance analyses observed among the seven populations of G. morbida isolates can reflect the history of the recent spread, and support the hypothesis of two separate pathogen introductions into the native region of black walnut—one into TN subpopulations and one into the Great Smoky Mountains National Park (GRSM population). Kolarik et al. (2011) indicated that the pathogen presence in the western US was unlikely to be a recent introduction, suggesting that a relationship between G. morbida and P. juglandis has been established for a significant amount of time outside the range reported by Wood and Bright (1992) (Kolarik et al. 2011). Nonetheless, it is important to note that black walnut trees are established but not native in the western US and therefore, host–pathogen interaction can respond differently to disease pressure. However, in the native range, anthropogenic movement of timber from infested locations and high susceptibility of black walnut (Utley et al. 2013) can support the hypothesis of relatively recent introduction of the pathogen that existed undetected for number of years. Also, initial disease symptoms resembled drought damage and therefore may have been undetected for number of years.
P. juglandis distribution is not continuous within the geographic range they inhabit (Seybold et al. 2012), which can provide an insight into a stochastic form of disease spread in the eastern US. This can be explained by several major factors including the presence of susceptible host species (Juglans spp.) and human-mediated movement of firewood, barked wood products and mulch from infested locations (Seybold et al. 2012; Utley et al. 2013). Our preliminary data support the hypothesis that TCD was unlikely to be spread via long distance dispersal of the pathogen, due to the restricted flight habit of P. juglandis. Unlike ophiostomatoid and ambrosia fungi, Geosmithia spp. do not have any known entomochoric adaptations for fungal spore dispersal (Kolarik et al. 2008). Currently, we have limited knowledge regarding any other beetle species involved in TCD complex spread, dissemination of spores using specialized organs for fungal transport such as mycangia, or mechanisms of pathogenicity and subsequent host responses. However, a more comprehensive population genetics study with larger sampling localities from the western US should elucidate the evolutionary history of both fungus and P. juglandis.
Cluster analysis indicated a high degree of genetic similarity among G. morbida isolates from TN subpopulations, which corresponds well to their geographical proximities and offers plausible explanation of the restricted flight habit of P. juglandis. Similar genetic comparisons were observed with other fungal pathogens (Ordoñez and Kolmer 2007; Stefansson et al. 2012). However, the grouping of OR-GRSM samples does not conclusively indicate that samples in the GRSM originated from Oregon, due to the limited number of isolates available in our study. Chronology records of G. morbida, spore dispersal and virulence, and overall pathogen relationship with both vector and host are still largely unknown.
Risk models proposed by McDonald and Linde (2002) indicated that asexual pathogens with high genotype flow represent higher risk for a disease outbreak due to a linked package of coadapted alleles. G. morbida does not have a known sexual state and as an asexual pathogen that does not recombine genes; there is a possibility for entire genotypes to be exchanged among populations (McDonald and Linde 2002). Subpopulations in the TN cluster exhibited high levels of gene flow and shared more genotypes when compared to the OR-GRSM cluster, which can increase the effective population size by increasing the size of the genetic neighborhood (McDonald and Linde 2002). In the case of G. morbida and findings of limited flight dispersal of P. juglandis, one plausible explanation that can affect the size of the genetic neighborhood is the anthropogenic movement of infested firewood into the native range of black walnut, resulting in higher gene/genotype flow than expected based on life history traits (McDonald and Linde 2002).
Expansion of non-native pathogen(s) into native forest communities can pose a significant risk due to the absence of prior co-evolutionary encounters and the evolutionary potential of the fungus (Tsui et al. 2012). Recent discoveries of TCD in the Great Smoky Mountains National Park (Hadziabdic et al. 2013) present a serious concern for forest health protection. To date, all confirmed cases of TCD have been in urban areas, along rural roadsides and/or fence rows. Recent report in GRSM is the first finding of G. morbida in a forest setting, which can cause a serious threat to conservation efforts of this native North American tree. GRMS is the highest visited national park in the country with more than 9.4 million recreational visits in 2010 (http://www.nps.gov) and human-mediated movement across US can be detrimental for disease establishment, spread and future outbreaks.
Future research should focus on understanding the biodiversity of both fungal and P. juglandis populations in infected black walnut communities in the eastern US compared to established fungal/insect populations in the western US where black walnuts are planted, but are not native. Understanding correlations between biology, host–pathogen–insect interactions, environmental influences and genetics in relation to pathogenicity of G. morbida will greatly improve our knowledge regarding disease epidemiology and provide necessary tools for control of potential disease outbreaks.
References
Amos W, Hoffman JI, Frodsham A, Zhang L, Best S, Hill VS (2007) Automated binning of microsatellite alleles: problems and solutions. Mol Ecol Notes 7:10–14
Becker DA, Brittingham MC, Goguen CB (2008) Effects of hemlock woolly adelgid on breeding birds at Fort Indiantown Gap, Pennsylvania. Northeast Nat 15:227–240
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cranshaw W, Tisserat N (2008) Pest alert: walnut twig beetle and thousand cankers disease of black walnut. Colorado State University. http://wci.colostate.edu/Assets/pdf/ThousandCankers.pdf
Elliott KJ, Swank WT (2008) Long-term changes in forest composition and diversity following early logging (1919–1923) and the decline of American chestnut (Castanea dentata). Plant Ecol 197:155–172
Eschtruth AK, Battles JJ (2008) Deer herbivory alerts forest response to canopy decline caused by an exotic insect pest. Ecol Appl 18:360–376
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10:564–567
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Grant JF, Windham MT, Haun WG, Wiggins GJ, Lambdin PL (2011) Initial assessment of thousand cankers disease on black walnut, Juglans nigra, in eastern Tennessee. Forests 2:741–748. doi:10.3390/f2030741
Hadziabdic D, Wadl PA, Vito LM, Boggess SL, Scheffler BE, Windham MT, Trigiano RN (2012) Development and characterization of sixteen microsatellite loci for Geosmithia morbida, the causal agent of thousand canker disease in black walnut (Juglans nigra). Conserv Genet Resour 4:287–289. doi:10.1007/s12686-011-9526-0
Hadziabdic D, Windham M, Baird E, Vito L, Cheng K, Grant J, Lambdin P, Wiggins G, Windham A, Merten P, Taylor G (2013) First report of Geosmithia morbida in North Carolina: the pathogen involved in thousand cankers disease of black walnut. Plant Dis. doi:10.1094/PDIS-06-13-0630-PDN
Hall B, Motzkin G, Foster DR, Syfert M, Burk J (2002) Three hundred years of forest and land-use change in Massachusetts, USA. J Biogeogr 29:1319–1335
Hansen MA, Bush E, Day E, Griffin G, Dart N (2011) Walnut thousand cankers disease alert, Virginia Tech-Virginia Cooperative Extension. http://www.vdacs.virginia.gov/plant&pest/disease-tcd.shtml
Hubisz M, Falush D, Stephens M, Pritchard J (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Res 9:1322–1332
Kolarik M, Kubatova A, Hulcr J, Pazoutova S (2008) Geosmithia fungi are highly diverse and consistent bark beetle associates: evidence from their community structure in temperate Europe. Microb Ecol 55:65–80
Kolarik M, Freeland E, Utley C, Tisserat N (2011) Geosmithia morbida sp. nov., a new phytopathogenic species living in symbiosis with the walnut twig beetle (Pityophthorus juglandis) on Juglans in USA. Mycologia 103:325–332
Langella O (1999) Populations 1.2.31. On web at: http://www.bioinformatics.org/~tryphon/populations/
Maliyakal EJ (1992) An efficient method for isolation of RNA and DNA from plants containing polyphenolics. Nucleic Acids Res 20:2381
McDonald BA, Linde C (2002) Pathogen population genetics, evolutionary potential and durable resistance. Annu Rev Phytopathol 40:349–379. doi:10.1146/annurev.phyto.40.120501.101443
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Nunes CF, Ferreira JL, Fernandes MCN, Breves SS, Generoso AL, Soares BDF, Dias MSC, Pasqual M, Borem A, Cançado GMA (2011) An improved method for genomic DNA extraction from strawberry leaves. Cienc Rural 41(8):1383–1389
Ordoñez ME, Kolmer JA (2007) Simple sequence repeat diversity of a worldwide collection of Puccinia triticina from durum wheat. Phytopathology 97:574–583
Page RDM (1996) TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358. doi:10.1093/bioinformatics/12.4.357
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetics software for teaching and research—an update. Bioinformatics. First published online July 20, 2012. doi:10.1093/bioinformatics/bts460
Penn State-Penn State Cooperative Extension (2011) http://extension.psu.edu/greenindustry/news/2011/housand-cankers-disease-tcd-detected-in-pa
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Pritchard JK, Donnelly P (2001) Case–control studies of association in structured or admixed populations. Theor Popul Biol 60:227–237
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Seybold SJ, Coleman TW, Dallara PL, Dart NL, Graves AD, Pederson LA, Spichiger SE (2012) Recent collecting reveals new state records and geographic extremes in the distribution of the walnut twig beetle, Pityophthorus juglandis Blackman (Coleoptera: Scolytidae), in the United States. Pan-Pacific Entomologist 88:277–280
Smouse PE, Long JC, Sokal RR (1986) Multiple regression and correlation extensions of the Mantel test of matrix correspondence. Syst Zool 35(4):627–632
Stefansson TS, Serenius M, Hallsson JH (2012) The genetic diversity of Icelandic populations of two barley leaf pathogens, Rhynchosporium commune and Pyrenophora teres. Eur J Plant Pathol 134:167–180. doi:10.1007/s10658-012-9974-8
Tisserat N, Cranshaw W, Leatherman D, Utley C, Alexander K (2009) Black walnut mortality in Colorado caused by the walnut twig beetle and thousand cankers disease. Plant Health Prog. doi:10.1094/PHP-2009-0811-01-RS
Tsui CK, Roe AD, El-Kassaby YA, Rice AV, Alamouti SM, Sperling FA, Cooke JE, Bohlmann J, Hamelin RC (2012) Population structure and migration pattern of a conifer pathogen, Grosmannia clavigera, as influenced by its symbiont, the mountain pine beetle. Mol Ecol 21:71–86
USDA-APHIS (2009) Pathway assessment: Geosmithia sp. and Pityophthorus juglandis Blackman movement from the western into the eastern United States. http://www.tn.gov/agriculture/publications/regulatory/tc_pathwayanalysis.pdf
USDA-Forest Service (FS) and Plant Protection and Quarantine (PPQ) (2012) Thousand cankers disease Survey Guidelines for 2012. http://www.aphis.usda.gov/plant_health/plant_pest_info/tcd/downloads/TCDSurveyGuidelines2012.pdf
Utley C, Nguyen T, Roubtsova T, Coggeshall M, Ford TM, Grauke LJ, Graves AD, Leslie CA, McKenna J, Woeste K, Yaghmour MA, Cranshaw W, Seybold SJ, Bostock RM, Tisserat N (2013) Susceptibility of walnut and hickory species to Geosmithia morbida. Plant Dis 97:601–607
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis DGM, Sninsky J, White T (eds) PCR protocols: a guide to methods and applications. Academic Press, San Diego, pp 315–322
Wood SL, Bright DE (1992) A catalog of Scolytidae and Platypodidae (Coleoptera) 2. Taxonomic index. Great Basin Nat Mem 13:1–1533
Wright S (1969) Evolution nad the genetics of populations. Vol. II. The theory of gene frequencies. University of Chicago Press, Chicago
National Park Service, Great Smoky Mountains. http://www.nps.gov/grsm/parkmgmt/statistics.htm
Acknowledgments
The authors thank the United States Department of Agriculture-ARS (Grant number 58-6404-1-637), United States Forest Service (Grant number 13-DG-11083150-033) and United States Forest Service-Special Technology Development Program (Grant numbers 13-DG-11083150-039) for financial support. The authors thank independent reviewers for their comments and suggestions for manuscript improvement. Special thanks to undergraduate research assistants Dixie Daniels and Matthew Aldrovandi for their laboratory help, and Dana Rhodes (Department of Agriculture, PA), Richard Baird (Mississippi State University) and Qunkang Cheng (University of Tennessee) for collecting and obtaining additional samples for our study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann.
Rights and permissions
About this article
Cite this article
Hadziabdic, D., Vito, L.M., Windham, M.T. et al. Genetic differentiation and spatial structure of Geosmithia morbida, the causal agent of thousand cankers disease in black walnut (Juglans nigra). Curr Genet 60, 75–87 (2014). https://doi.org/10.1007/s00294-013-0414-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-013-0414-x