
Abstract
A monooxygenase gene was isolated from a biocontrol strain of Trichoderma hamatum and its role in biocontrol was investigated. The gene had homologues in other fungal genomes, but was not closely related to any fully characterised gene. The T. hamatum monooxygenase gene was expressed specifically in response to the plant pathogens Sclerotinia sclerotiorum, Sclerotinia minor and Sclerotium cepivorum, but not in response to Botrytis cinerea or T. hamatum. Expression of the gene did not occur until contact had been made between the two fungal species. Homologues in T. atroviride and T. virens showed similar expression patterns. Expression of the gene in response to S. sclerotiorum was influenced by pH, with a peak of expression at pH 4, and was subject to nitrogen catabolite repression. Disruption of the monooxygenase gene did not affect the growth or morphology of T. hamatum, but caused a decrease in its ability to inhibit the growth and sclerotial production of S. sclerotiorum. The monooxygenase gene had a role in the antagonistic activity of Trichoderma species against specific fungal plant pathogens and is therefore a potentially important factor in biocontrol by Trichoderma species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Trichoderma species are effective as biological control agents for a range of crop diseases caused by fungal plant pathogens. Their use has the potential to reduce considerably the quantity of chemical fungicides used in agriculture; however, greater consistency is required for widespread acceptance. Exploration of the molecular basis of biocontrol interactions has the potential to yield strategies for improving the reliability of biocontrol by Trichoderma species.
Trichoderma biocontrol agents act through a number of mechanisms such as mycoparasitism, competition, antibiosis (Hjeljord and Tronsmo 1998) and induction of systemic resistance (Meyer et al. 1998), or combinations of these mechanisms (Lorito et al. 1996). As yet, only mycoparasitism has been extensively investigated at a molecular level. Several cell wall degrading enzymes are involved in the mycoparasitic attack on the host fungus including proteases, chitinases and glucanases, and the genes encoding some of these have been well characterised (Geremia et al. 1993; Carsolio et al. 1994; Donzelli et al. 2001). Increased expression of genes encoding cell wall degrading enzymes, such as the alkaline protease prb1, endochitinase chit 33 and endoglucanase egl1, can improve biocontrol (Flores et al. 1997; Migheli et al. 1998; Limon et al. 1999; Pozo et al. 2004), but this is not always the case, as with the endochitinase gene ech42 (Carsolio et al. 1999). Other genes that appear to have a role in mycoparasitism have also been identified, such as a putative amino acid permease (Vasseur et al. 1995) and the tga1 and tga3 genes encoding G-protein α-subunits (Rocha-Ramirez et al. 2002; Zeilinger et al. 2005). Although Trichoderma species are known to produce a range of antibiotics, little is known about the genes involved in their synthesis (Howell 1998). Molecules involved in the induction of systemic resistance in plants by Trichoderma species are currently being characterised (Hanson and Howell 2004; Harman et al. 2004). Induction of systemic resistance in plants by Trichoderma asperellum involves the jasmonic acid pathway (Shoresh et al. 2004), but the details of this process remain to be elucidated.
Commercially available Trichoderma preparations for biocontrol are usually Trichoderma atroviride or Trichoderma harzianum. A less well-known species, Trichoderma hamatum, is also an effective biocontrol agent against Sclerotinia sclerotiorum, Sclerotinia minor, Botrytis cinerea, Pythium ultimum and Rhizoctonia solani (Harman et al. 1980; Rabeendran 2000; Horst et al. 2005). The interaction between T. hamatum and S. sclerotiorum involves increased expression of the T. hamatum homologues of the ech42 and prb1 genes from T. atroviride, which encode host cell wall-degrading chitinase and proteinase enzymes (Steyaert et al. 2004). A number of other T. hamatum genes, which showed increased expression during interaction with S. sclerotiorum, were identified using suppressive subtractive hybridisation (Carpenter et al. 2005). In the present study, we report on the characterisation of one of those genes, a monooxygenase, and investigate its role in biological control by T. hamatum.
Materials and methods
Strains
T. hamatum LU593, T. atroviride LU132 and T. virens LU555 were identified by morphology and ITS1 sequencing (Steyaert et al. 2004). S. sclerotiorum, S. minor, B. cinerea, Sclerotium cepivorum and P. ultimum were identified by morphology. Cultures were grown on potato dextrose agar (PDA) at 22°C in the dark. PDA for culturing P. ultimum was amended with 0.5% chloramphenicol.
Genomic DNA isolation
Trichoderma spp. were cultured in potato dextrose broth (PDB) in 6 ml Petri dishes for 3 days in the dark at 22°C. Mycelium was lifted out of the medium, rinsed in sterile water and squashed between paper towels to remove liquid. The mycelium was ground in liquid nitrogen, and DNA was extracted using a PureGene kit (Gentra Systems, Minneapolis, MN). Alternatively, when isolating small quantities of DNA for PCR, mycelium was scraped from 3- to 5-day-old PDA plates, ground in lysis buffer using a micro-pestle and purified using the PureGene kit.
Genomic library construction and screening
A T. hamatum genomic library was constructed by partially digesting LU593 genomic DNA with Sau3AI and isolating the 8- to 12-kb fragments from an agarose gel. The Sau3AI fragments were ligated into a Zap Express vector (Stratagene, La Jolla, CA) cut with BamHI. The library was packaged, amplified and stored at −80°C.
The T. hamatum genomic library was plated out at 4 × 104 per 100 cm2 plate in E. coli XL1-BlueMRF’ cells. Plaque lifts were done using Hybond-N+ (Amersham Biosciences, UK) and fixed using 0.5 M NaOH followed by a wash in 5× SSC. The library was screened with a 330-bp fragment of a monooxygenase gene (denoted G3) previously isolated from a subtraction library (Carpenter et al. 2005) and using ECL labelling and detection (Amersham Biosciences). Three rounds of screening were done to ensure that the clone was pure. The pBK-CMV phagemids with inserts were excised from the lambda vector as per the Zap Express kit instructions and plasmid DNA was isolated.
Isolation of homologous genes from other Trichoderma species
Genes homologous to those found in T. hamatum were amplified from T. atroviride and T. virens using degenerate primers. To amplify within the coding region, primers G3degf (5′-GTSGCCATCATYGGYGCNGG-3′) and G3degr2 (5′-TCRAARGCCTGGTTSACRCCYTG-3′) were used. To amplify 5′ of the monooxygenase gene in T. atroviride, primer LU132G3forprom (5′-TAGAGCTTCTCAAAGTGATG-3′) from the T. atroviride coding sequence and degenerate primer acetyltransdeg (5′-ATYCARATGCACATYGACTGCAT-3′) from the coding region of the gene upstream of the monooxygenase were used.
Sequence analysis
Plasmids were sequenced using ABI BigDye terminator chemistry and an ABI3100 sequencer (Applied Biosystems, Foster City, CA). Sequencing primers were designed using Primer 3 (http://Frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) with a melting temperature of 50°C. Both strands were sequenced and assembled using Sequencher 4.2 (Gene Codes, Ann Arbor, MI). Sequence data was analysed using Blast searches of GenBank and fungal genomes. Promoter analysis was done using DNAman 4.0 (Lynnon Biosoft, Quebec, Canada) to search for regulatory motifs previously found to be associated with mycoparasitism and/or biocontrol (Steyaert et al. 2003).
Confrontation assays and RNA extraction
Minimal medium agar (MMA) (Carsolio et al. 1994) containing 0.2% glucose was covered with a sheet of sterile, lightly sandpapered cellophane and inoculated with two agar plugs 4.5 cm apart. Confrontation plates were inoculated with a Trichoderma species and a plant pathogen, whereas control plates received two plugs of Trichoderma spp. Plates were incubated at 22°C in the dark for 2–4 days.
The mycelium was harvested from the interaction zone of the two colonies by gently scraping it off the cellophane using a scalpel blade. It was immediately frozen in liquid nitrogen and ground using a mortar and pestle. Total RNA was isolated using Trizol Reagent (Invitrogen, Carlsbad, CA), resuspended in water and stored at −80°C.
Northern and Southern blotting
RNA was separated by electrophoresis in a 1.2% formaldehyde–agarose–MOPS gel (Sambrook and Russell 2001). Genomic DNA (5 μg) was separated in a 0.8% agarose Tris–acetate–EDTA (TAE) gel. Nucleic acids were transferred to Hybond-N+ membrane (Amersham Biosciences) by capillary transfer. Blots were probed using DIG chemiluminescent labelling (Roche, Mannheim, Germany) according to the manufacturer’s recommendations.
Gene replacement construct
A gene replacement construct was created using overlap (fusion) PCR (McPherson 2000) to join approximately 2.5 kb from either flank of the G3 monooxygenase coding region to a hygromycin resistance cassette. The upstream flank was amplified from a pBK-CMV-G3 phagemid using primers G3pf2, which has a HindIII site at the 5′-end, and G3pr (Table 1). The downstream flank was amplified using G3tf and G3tr2, which has a KpnI site at the 5′-end. The hygromycin resistance (hph) cassette from pAN7-1 (Punt et al. 1987), including Aspergillus nidulans gpd promoter and trpC terminator, was amplified using primers hygf2 and hygr2 in which the 5′-ends are homologous to primers G3tf and G3pr, respectively. The components of the replacement construct were amplified using the Expand Long Template PCR system (Roche, Mannheim, Germany) and purified from a 1% agarose gel. The three segments were joined and amplified in a second PCR, using primers G3pf 2 and G3tr2 with 120 ng of template made up of the three segments in equimolar amounts. The overlap fragment was digested with KpnI and HindIII and cloned into the binary vector p0300 that was also digested with KpnI and HindIII. The plasmid p0300 was made from pCAMBIA 2300 (CAMBIA, Canberra, Australia) by digestion with BstXI and XhoI to remove the NPTII coding region and CaMV35S promoter, producing a binary vector in which the only sequences between the left and right borders was the multiple cloning site (MCS) in the lacZα gene and the redundant CaMV35S terminator. The gene replacement construct (pG3KO2) was checked by sequencing across each junction. The plasmid pG3KO2 was then transformed into Agrobacterium tumefaciens strain EHA105 by electroporation and transformants were selected on Luria broth (LB) agar containing 50 μg/ml kanamycin and 10 μg/ml rifampicin.
Agrobacterium-mediated transformation of T. hamatum LU593
T. hamatum LU593 was transformed by pG3KO2 using the method of Zeilinger (2004). The A. tumefaciens EHA105 containing pG3KO2 was grown at 24°C in induction medium with 200 μM acetosyringone until the OD at 660 nm was 0.2. Agrobacterium cultures (500 μl) were mixed with 107 conidia, spread on sterile cellophane placed on induction medium agar and incubated for 48 h at 22°C. The cellophane was then transferred to PDA supplemented with 300 μg/ml hygromycin and 200 μg/ml timentin, and overlaid with 4 ml 1/2x PDA with 300 μg/ml hygromycin and 200 μg/ml timentin at 42°C. Transformants were subcultured twice on to PDA containing hygromycin and timentin, and DNA was purified from mycelium scraped from the second subculture. Transformants were checked for the presence of a gene replacement by PCR using primers kotest1 and kotest2, which were designed to give a 2.7 kb product in a knockout and no product in the wild-type, and primers kotest 1 and kotest3, which produced a 6.7 kb product in knockouts and 4 kb in the wild-type. Transformants carrying the gene knockout were purified by two rounds of single spore isolation. A Southern blot was used to confirm the presence of a gene knockout and absence of additional ectopic integration. DNA from the knockout mutants was digested with PstI, and the Southern blot was probed with two fragments of 550 and 1 kb amplified from pG3KO2 using primers hygcodf and hygcodr and G3pf and G3pr, respectively.
Phenotypic characterisation of knockout mutants
The growth rate and morphology of the knockout mutants on PDA was compared to that of the wild-type LU593. Strains were subcultured onto PDA in triplicate and incubated at 22°C. The colony radius was measured daily for 3 days, and appearance of the colony including the presence of spores was noted at 8, 10 and 15 days after inoculation.
Metabolism of salicylate was determined by two methods. Salicylate fluoresces under UV transillumination (312 nm) and this fluorescence can be detected in agar plates. Catechol, the product of salicylate metabolism, is not detected (Ishiyama et al. 2004). MMA containing 0.1% salicylate was inoculated with LU593 or one of the knockout mutants, and incubated at 22°C. Plates were photographed using broad bandwidth UV transillumination (290–365 nm). Salicylate metabolism was also determined by measuring biomass produced with salicylate as a sole carbon source. Spores (105) from LU593 and knockout mutants were grown in 100 ml MM with 2.5 mM salicylate, or 2.5 mM glucose or no carbon source at 22°C and 200 rpm. After 9 days of growth, the cultures were filtered, dried and weighed.
Confrontation assays for evaluating knockout mutants were conducted by inoculating MMA or PDA plates with agar plugs of S. sclerotiorum and LU593 or a knockout mutant, 4 cm apart, and incubating at 22°C. The growth and appearance of the colonies were recorded every few days for 23 days. Then the sclerotia produced on each plate were collected, counted, dried and weighed.
The biocontrol ability of the knockout mutants relative to the wild-type LU593 was determined using a cabbage leaf midrib assay (Rabeendran 2000). The test strains and S. sclerotiorum were cultured on PDA for 4 days, and 10 mm discs cut from the plates using a cork borer. A S. sclerotiorum disc was placed in the centre of a Petri dish on moistened sterile sand with its mycelium side up beneath a Trichoderma disc placed with its mycelium side down. The midribs were excised from cabbage leaves, sterilised with 1% sodium hypochlorite for 3 min, then washed 3× with sterile water. The ends were trimmed and the midribs cut into 30-mm sections. Three pieces of cabbage midrib were placed in each Petri dish radiating outwards from the fungal discs, ensuring contact between the cabbage and agar. Controls included each strain paired with a disc of sterile PDA. Ten replicates were used for S. sclerotiorum alone and paired with each T. hamatum strain. Controls, consisting of each T. hamatum strain alone or sterile PDA, were replicated six times. The assays were incubated at 22°C. At 4 and 7 days after inoculation, the proportion of the cabbage midrib that was softened and discoloured was determined using sterile forceps. After 21 days, any sclerotia produced were counted, dried and weighed. The results were analysed by ANOVA using Genstat 9.
Creation of GFP promoter fusion mutants
A binary vector for constitutive expression of GFP (p0300GFP) was constructed by digesting pCT74 (Lorang et al. 2001) with EcoRI and KpnI to remove the hph and GFP cassettes on a single fragment, which was then cloned into the MCS of p0300. The G3 monooxygenase promoter fusion vector (pG3GFP) was constructed by cutting p0300GFP with NcoI and SalI, which removed both the ToxA promoter from the GFP coding region and the hygromycin resistance cassette. The G3 gene promoter was amplified by PCR using Pfu polymerase (Fermentas) using primers 593G3promf2 (5′-ATATGTCGACAAGCAGCTAGTTGCGCCTGT-3′) and 593G3promr2 (5′-ATACCATGGTTTGGGGTTTTTGTTGTGAC-3′), which incorporated restriction sites for the enzymes SalI and NcoI. The product was cut with SalI and NcoI and ligated into the MCS of p0300GFP cut with the same enzymes.
The GFP plasmids p0300GFP and pG3GFP were sequenced to check that no errors had been introduced into the G3 promoter sequence or GFP coding sequence and that the structure was correct. The vectors were then transformed into A. tumefaciens EHA105 by electroporation. T. hamatum LU593 was transformed with p0300GFP, and also cotransformed with pG3GFP and pYT6 in a 2:1 ratio, using Agrobacterium-mediated transformation. pYT6 is a binary vector containing the hygromycin B resistance (hph) gene under the control of the Aspergillus glaA promoter and trpC terminator.
Induction of the G3 monooxygenase promoter in GFP fusion mutants
Spores (10,000) from wild-type LU593 or the transformed strains carrying constitutively expressed GFP and GFP controlled by the G3 monooxygenase promoter were germinated and grown in 250 μl PDB, in a bioimaging microplate with black walls and a clear bottom to the wells (BD Biosciences, Palo Alto, CA). After 5 days at 22°C, the PDB was removed by pipetting and 100 μl of test substance(s) added. Incubation at 22°C was resumed. Fluorescence and absorbance were measured on each of the following 4 days using a Fluostar microplate reader (BMG Labtech, Offenburg, Germany) with excitation at 485 nm and emission at 520 nm for detection of GFP, and using excitation at 590 nm for absorbance readings. S. sclerotiorum was cultured in PDB and treated in the following ways to produce reagents to test for stimulation of the G3 gene promoter. For live S. sclerotiorum, the culture (including broth) was fragmented using a blender. For autoclaved mycelium, the mycelium was separated from medium by vacuum filtration using a 0.22-μm filter, then blended in water and autoclaved. The filtrate was also tested, as were concentrated fractions of the filtrate obtained using Centriprep centrifugal filter devices (Millipore, Bedford, MA) with molecular weight cut-offs of 50,000 and 10,000. Purified cell walls were prepared from mycelium by grinding in liquid nitrogen and repeated washes with distilled water (Geremia et al. 1993). Chitin was suspended in water at a concentration of 1% and autoclaved. The effect of pH was determined using citrate–phosphate (McIlvaine) buffers at pH 3–7. The effect of glucose and ammonium availability was determined using glucose at concentrations of 2.5, 0.5 and 0.1%, and ammonium acetate at 1.0 and 0.1%. Three replicates were used and results were analysed by ANOVA.
Results
Monooxygenase gene sequence analysis
Screening of the T. hamatum genomic library with SSH clone G3 yielded a clone with a 7.8 kb insert, which contained the complete monooxygenase gene (GenBank accession EU124654). The gene consisted of a single exon encoding a protein of 410 amino acids, which had 64% identity to a monooxygenase AF110 from Aspergillus flavus (GenBank accession AAT65716) (Zhang et al. 2004). The protein sequence also showed high similarity to proteins predicted from sequences from other Aspergillus species and Gibberella zeae (Table 2). Comparison with the Fusarium graminearum (Gibberella zeae) genome (http://mips.gsf.de/genre/proj/fusarium) yielded more than 30 sequences with significant homology (E value < e−05). Of these, one stood out as having much greater similarity than the rest (1.4e−129) suggesting it was the homologous gene. Some of these proteins with the greatest similarity to the G3 monooxygenase have been predicted to be salicylate hydroxylases, but this role has not been confirmed experimentally. Comparison was therefore made with proteins whose functions have been confirmed. The G3 sequence was compared to the sequences of Agaricus bisporus 4-aminobenzoate hydroxylase, Saccharomyces kynureneine 3-monooxygenase, two bacterial salicylate hydroxylases and a bacterial 3-hydroxybenzoate hydroxylase. Although all had significant E values, all showed low identity (14–18%), indicating that they are related to G3, but not closely (Table 2).
Alignment of the T. hamatum monooxygenase amino acid sequence with that of A. flavus AF110 revealed conserved residues involved in FAD binding (Fig. 1). The Rossman fold motif GXGXXG, which binds the ADP moiety of FAD was present close to the N-terminus. Towards the C-terminus there was a conserved region, which included the GD motif that binds the ribose moiety of FAD. The DG motif described in flavoprotein hydroxylases by Eppink et al. (1997) was also present. Comparison of the G3 monooxygenase sequence to the most similar fungal sequences revealed an additional motif with the conserved sequence DIGGA, which was not present in the more distantly related sequences. It was present only in the most similar sequence from each fungal species, including the first four listed in Table 2. This motif corresponded in position to the GG motif described by Vallon (2000); however, the amino acids surrounding the core GG were different.

Alignment of the T. hamatum G3 monooxygenase amino acid sequence with the Aspergillus flavus homologue. Identical residues are indicated by vertical lines and conservative substitutions are indicated by dots. Asterisks indicate the conserved residues involved in binding FAD. The first box represents the region that binds the ADP moiety of FAD and includes the Rossman fold motif GXGXXG. The second box indicates the GG motif (Vallon 2000). The third box indicates the DG motif conserved in flavoprotein hydroxylases (Eppink et al. 1997), and the fourth box shows the GD motif that binds the ribose moiety of FAD
Sequencing 5′ of the G3 monooxygenase coding region revealed a 769 bp intergenic region and a predicted adjacent gene with homology to acetyl transferases. Sequencing 5′ of the acetyl transferase gene revealed homology to another unidentified fungal protein (Fig. 2). Sequencing 3′ of the stop codon of the G3 monooxygenase revealed 642 bp before the predicted stop codon of the next gene. The downstream gene was identified by sequence homology as the delta subunit of the coatomer complex (δ-COP), also known as the archain gene, and this gene extended to the end of the clone, which encompassed the complete δ-COP gene. The arrangement of the acetyl transferase, monooxygenase and δ-COP genes showed synteny with the T. reesei genome (http://genome.jgi-psf.org/Trire2/Trire2.home.html); however, upstream of the acetyl transferase, the synteny is broken as T. reesei that has a retroelement and a hypothetical protein unrelated to the one in T. hamatum (Fig. 2).

Arrangement of the genes in the genomic library clone carrying the T. hamatum G3 monooxygenase gene and in the equivalent region of the T. reesei genome. The G3 monooxygenase gene was flanked by genes predicted to be the delta unit of the coatomer protein and an acetyl transferase. 5′ of the acetyl transferase, the species differed, as T. hamatum had a hypothetical protein, which was present in a different part of the T. reesei genome, whereas T. reesei had a putative retroelement. Arrows show the direction of the open reading frames, which are represented by the grey boxes
G3 monooxygenase homologues in other species of Trichoderma
A part of the coding region of the G3 monooxygenase homologues from T. atroviride and T. virens were amplified and sequenced for use as probes so that the expression of the monooxygenase gene in these species could be compared to that of T. hamatum. The homologous genes from T. atroviride and T. virens were 87 and 81% identical, respectively, to T. hamatum over the 898 bp sequenced, and were 83% identical to each other. A region of 1.6 kb 5′ to the T. atroviride monooxygenase coding sequence was also amplified and sequenced so that the promoter sequences of the two species could be compared. A putative acetyl transferase gene was present 5′ of the monooxygenase gene in T. atroviride as was previously found in T. hamatum. In T. atroviride, the predicted stop codon of the acetyl transferase was 967 bp 5′ of the monooxygenase start codon, whereas in T. hamatum the predicted coding regions of the two genes are separated by 769 bp.
Analysis of the G3 monooxygenase gene promoter region
The promoter regions of the T. hamatum and T. atroviride G3 monooxygenase genes were searched for motifs known to be involved in the regulation of genes associated with mycoparasitism and biocontrol. For this purpose, the promoter region was defined as extending from the putative stop signal of the upstream acetyl transferase gene to the monooxygenase start codon, as the transcription start point had not been determined. Both the T. hamatum and T. atroviride G3 promoters had sequences matching 11 of the motifs searched for, specifically, the TATA box, CCAAT box, GATA, CreA, STRE, PacC, BrlA, AbaA, AceI, AceII and MYC1(Cortes et al. 1998; Steyaert et al. 2003). In most cases, the positions of the motifs were not conserved between the sequences from the two Trichoderma species, and as they are generally short motifs, some of which would be expected to occur randomly within these sequences, many may not be functional regulatory elements. The mycoparasitism elements MYC2–4 were not present in either sequence. There were a few motifs whose positions were conserved between the two promoters (Fig. 3) and were therefore considered more likely to be functional. CCAAT and PacC (pH regulation) motifs occurred in close proximity (26–31 bp between them) lying 240–280 bp upstream of the start codon. GATA (nitrogen repression) and BrlA (sporulation) motifs occurred at the 5′-end of the promoters, separated by 16–26 bp and lying within 70 bp of the preceding stop codon. Additionally, both sequences had MYC1 (mycoparasitism) elements positioned approximately 110 bp apart. Comparison of the two promoter sequences also revealed an overall similarity of 66% and a deletion in the middle of the T. hamatum promoter relative to the T. atroviride promoter. Alignment of the sequences revealed six blocks of identity ranging in size from 10 to 30 bp, which may represent novel regulatory sequences (Fig. 3). One of these contained a conserved CreA (carbon repression) motif. Smaller identical segments also occurred.

Comparison of the promoter regions of the T. hamatum and T. atroviride G3 monooxygenase genes. The black bars represent the sequence from the stop codon of the predicted upstream gene to the start codon of the monooxygenase. The dashed line represents a deletion in the T. hamatum sequence. Rectangles represent blocks of 100% identity with the size of the block in base pairs shown below. Regulatory motifs known from other genes are shown only when the position was conserved between the two species, and are depicted as follows: black diamond GATA, white diamond BrlA, arrow MYC1, triangle CreA, white star CCAAT, black star PacC
The T. hamatum G3 promoter was also compared to that of T. hamatum chit42 (ech42) and prb1, two biocontrol genes predicted to be regulated by common pathways, and to the T. reesei G3 homologue, but notable similarities were not observed.
Southern blot
Southern analysis produced a single band in each of three restriction digests (EcoRI, HindIII, PstI), which showed the G3 monooxygenase gene to be a single copy gene. Single bands were also produced by HindIII digests of T. atroviride and T. virens DNA showing the gene to be present in these species also (data not shown).
Expression of the T. hamatum G3 monooxygenase over time
Northern analysis revealed that the G3 monooxygenase was not expressed in T. hamatum grown alone or in confrontation with itself, but was expressed in confrontation with S. sclerotiorum. Expression of the gene did not occur until the two colonies were overlapping (Fig. 4). The lack of expression when T. hamatum is grown in confrontation with itself showed that the expression is specific to interaction with S. sclerotiorum and not simply a stress response induced by starvation.

Northern analysis showing expression of T. hamatum G3 monooxygenase in T. hamatum grown alone, grown in confrontation with itself and grown in confrontation with S. sclerotiorum. Day 2 was 2 days after inoculation and prior to contact of the two colonies on confrontation plates. On day 3, colonies had just made contact and overlapped by 1–3 mm. On day 4, they overlapped by approximately 10 mm. The top panel was probed with monooxygenase gene, and the lower panel shows ethidium bromide-stained RNA as a loading control
Induction of expression by alternative host species
The confrontation assay was repeated using other plant pathogen species and using T. atroviride and T. virens. Expression of the T. hamatum G3 monooxygenase gene in confrontation with S. cepivorum was at a level similar to that induced by S. sclerotiorum (Fig. 5a). S. minor and P. ultimum induced the monooxygenase to a lesser extent, barely detectable in the case of P. ultimum. No monooxygenase expression was detected in response to B. cinerea. Expression of the monooxygenase homologue in T. atroviride and T. virens followed a similar pattern (Fig. 5b, c). In these species, expression was induced by S. sclerotiorum and S. cepivorum but not by B. cinerea nor in the absence of a host species.

Northern analysis of G3 monooxygenase expression in Trichoderma spp. in response to different host species. Top panels were probed with the G3 monooxygenase gene from the respective Trichoderma species. Lower panels were probed with 18s as a loading control. RNA was extracted when colonies were overlapping by 5–10 mm. Th T. hamatum, Ta T. atroviride, Tv T. virens, Ss S. sclerotiorum, Sm S. minor, Sc S. cepivorum, Bc B. cinerea, Pu P. ultimum, — Trichoderma spp. grown alone
Induction of expression of the G3 monooxygenase determined using GFP promoter fusion mutants
Several transformants, which carried the constitutively expressed GFP and GFP regulated by the G3 promoter, were obtained. One of each was chosen for analysis of gene expression based on the strength of GFP expression and the similarity of growth and morphology to the wild-type.
The expression of the G3 monooxygenase gene in response to live S. sclerotiorum was significantly influenced by pH (P < 0.01). Two days after the addition of citrate–phosphate buffers, when the highest level of expression occurred, G3 expression in the presence of S. sclerotiorum was greatest at pH 4. Both pH 3 and 4 gave greater expression than in unbuffered conditions, and expression decreased as pH increased from 5 to 7 (Fig. 6a). The expression of G3 was also significantly affected by ammonium ion concentration (P < 0.01) when measured after 3 days. Expression was induced by live S. sclerotiorum when ammonium acetate had been added at 0 and 0.1%, but not at 1% (Fig. 6b). The concentration of glucose did not significantly affect G3 expression. The three glucose concentrations (0.1, 0.5 and 2.5%) and no glucose all gave comparable expression of G3 in response to live S. sclerotiorum. In the absence of S. sclerotiorum, there was no expression of G3 at any pH or ammonium ion or glucose concentration. There was no induction of gene expression in response to chitin, or any of the fractions derived from S. sclerotiorum other than the live S. sclerotiorum culture.

G3 monooxygenase gene expression induced by S. sclerotiorum is influenced by pH and nitrogen availability. a The fluorescence produced by wild-type LU593 and a G3 promoter fusion mutant 2 days after the addition of citrate-phosphate buffers with or without live, blended S. sclerotiorum. The monooxygenase gene was induced only in the presence of S. sclerotiorum, and induction was greatest at pH 3 and 4 or in unbuffered medium. b The fluorescence produced by the same strains 3 days after the addition of 0, 0.1 and 1.0% ammonium acetate with or without live, blended S. sclerotiorum. The monooxygenase gene was induced only in the presence of S. sclerotiorum and at low or zero levels of ammonium acetate. Gene expression was repressed in the presence of 1% ammonium acetate. Error bar = ±standard error of the mean. wt wild-type LU593, G3 G3 promoter:GFP fusion mutant, Ss S. sclerotiorum
Identification of gene knockout mutants by PCR and Southern blotting
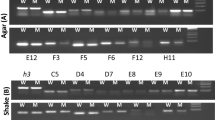
Screening of 30 putative knockout transformants by PCR revealed three mutants, which generated PCR products indicative of a gene knockout. After purification by single spore isolation, Southern blot analysis of these three mutants, using a restriction enzyme which cut within the hygromycin gene, showed that the wild-type fragment was replaced by two smaller fragments, confirming that all three mutants were knockout mutants (Fig. 7.). The Southern blot also confirmed that no additional ectopic integrations had occurred in any of the knockouts.

Southern blot showing disruption of the G3 monooxygenase gene in knockout mutants. DNA was digested with PstI, and the blot was probed with two fragments from the coding region of the hygromycin gene and the promoter region of the G3 gene, respectively. A PstI site was present in the hygromycin gene-coding region. A 9-kb fragment in the wild-type was replaced by two smaller fragments (5 and 6 kb) in the knockouts (KO13, KO15, KO24), and no additional ectopic integrations were evident. KO32 showed the 9-kb wild-type fragment and an ectopic integration
Phenotypic characterisation of G3 monooxygenase gene knockout mutants
The growth rate of the knockout mutants on PDA was not significantly different from that of the wild-type, consistent with a lack of gene expression during normal growth. The appearance of the colonies was also similar to that of the wild-type, except for a possible small reduction in sporulation. Neither of the assays for salicylate metabolism showed a difference between wild-type LU593 and the G3 knockout mutants, indicating that salicylate is unlikely to be the substrate for the monooxygenase enzyme.
The behaviour of the knockout mutants in confrontation with S. sclerotiorum was compared to LU593 on PDA and on MMA. In all cases, the T. hamatum strains and S. sclerotiorum grew rapidly until the two colonies met. Then S. sclerotiorum ceased to grow, while T. hamatum slowly overgrew S. sclerotiorum. On PDA, LU593 overgrew S. sclerotiorum to a greater extent than any of the knockout mutants; however, the difference was only significant for KO13 (P < 0.05) (Table 3). On MMA, LU593 appeared to overgrow S. sclerotiorum more quickly than any of the knockouts; however, the sparse growth produced on this medium by both species made it difficult to distinguish them with certainty. The number and total weight of sclerotia produced on each PDA confrontation plate were recorded. The number and weight of sclerotia produced in the presence of the knockout mutants were greater than those for the wild-type, indicating that the knockouts had a decreased inhibitory activity against S. sclerotiorum (Table 3). However, this difference was only statistically significant (P < 0.05) in the case of KO13 (number and weight) and KO15 (number only). Overall, the gene disruption caused a small decrease in the antagonistic activity of T. hamatum against S. sclerotiorum.
Two of the knockout mutants, KO13 and KO24, were tested for their ability to control the rot of cabbage leaf midrib sections by Sclerotinia sclerotiorum. In both assays, the wild-type LU593 gave excellent control of the Sclerotinia disease, while the negative control showed complete decay of the cabbage tissue (Table 4). Biocontrol by KO13 was not significantly different from the wild-type for any of the parameters measured. However, biocontrol by KO24 was significantly less than the wild-type in both the proportion of cabbage tissue decayed 4 and 7 days after inoculation (P < 0.01), and in the number and weight of sclerotia produced (P < 0.01).
Discussion
A novel gene with potential importance in biocontrol was isolated from T. hamatum. Comparative sequence analysis showed the gene to encode a protein belonging to the large and diverse family of monooxygenases, which are involved in various aspects of cellular metabolism. Proteins, which are very similar to the G3 monooxygenase, have been predicted from fungal genome sequencing, but the function of these has not been determined. The G3 monooxygenase had the sequence motifs characteristic of a FAD-dependent monooxygenase (Eppink et al. 1997; Vallon 2000), but was not closely related to any well-characterised flavoprotein. Only distantly related proteins have been properly identified, and these include a range of monooxygenases such as an aminobenzoate hydroxylase from Agaricus (Tsuji et al. 1996), kynurenine 3-hydroxylase from Saccharomyces, and bacterial hydroxylases of salicylate (Bosch et al. 2000) and other benzoate derivatives (Liu et al. 2005). Therefore, sequence homology did not give a strong indication of the reaction catalysed by this enzyme.
Some of the genes showing greatest similarity were predicted to encode salicylate hydroxylases. This was of particular interest due to the association of salicylate with the induction of systemic resistance in plants by Trichoderma species (Harman et al. 2004) and also the ability of salicylate to induce expression of the endochitinase gene ech42 in T. atroviride (Lutz et al. 2004). Salicylate metabolism was tested using the gene disruption mutants; however, the results were not consistent with salicylate as a substrate; so, the substrate of this monooxygenase remains to be determined.
Sequencing of fungal genomes, such as Fusarium graminearum (http://mips.gsf.de/genre/proj/fusarium) and Neosartorya fischeri (http://tigrblast.tigr.org), has shown them to contain many genes encoding a variety of monooxygenases, most of which have not had their function determined. Using sequence similarity as an indication of function in a large gene family like this must be approached with caution, as a significant level of similarity (<e−5) does not necessarily indicate a common function.
Monooxygenase genes sometimes lie in clusters with other functionally related genes. The T. hamatum G3 monooxygenase gene was positioned between a putative acetyl transferase gene and the coatomer delta subunit (δ-COP) gene, an arrangement which was conserved in T. reesei (http://genome.jgi-psf.org/Trire2/Trire2.home.html). It seems unlikely that the δ-COP gene has functional relationship to the monooxygenase, as δ-COP is a highly conserved gene present in diverse eukaryotes, with a fundamental role in eukaryote cell biology concerning intracellular protein transport (Tunnacliffe et al. 1996). It is possible that the acetyl transferase and the adjacent hypothetical protein are functionally related to the monooxygenase; however, there is insufficient information known about these proteins to draw any conclusions. Upstream of the acetyl transferase, the synteny with T. reesei was broken. If these genes are functionally related, the break in synteny could reflect a functional difference between the two species, i.e., the monooxygenase gene may have a role in biocontrol in T. hamatum but a different role in T. reesei, which is not a recognised biocontrol species. AF110, the Aspergillus flavus gene that gave the highest similarity to the T. hamatum monooxygenase gene, is adjacent to a gene cluster, which synthesises the mycotoxin aflatrem and is likely to be a part of this gene cluster (Zhang et al. 2004). This could be an indication that the T. hamatum gene is involved in synthesis of a mycotoxin; however, there was no synteny between the T. hamatum genes and the A. flavus cluster.
The expression of the T. hamatum monooxygenase gene was stimulated specifically during interaction with S. sclerotiorum and certain other plant pathogenic fungi. Expression of the gene was not evident during growth on PDA and did not begin until contact was made between the two species. This strongly suggested that the gene product had a role in the antagonistic attack of Trichoderma spp. on other fungal species. Other genes involved in biocontrol, such as prb1, nag1 and chit42, showed similar patterns of expression (Carsolio et al. 1994; Cortes et al. 1998; Zeilinger et al. 1999; Steyaert et al. 2004). However, the expression of the G3 monooxygenase gene in response to plant pathogens was more specific than some of these other genes. For example, chit42 (ech42) showed a basal level of expression in the absence of a plant pathogen in both T. hamatum and T. atroviride (Carsolio et al. 1994; Steyaert et al. 2004), and was stimulated by light in T. atroviride (Carsolio et al. 1994). This was not the case for G3, which appeared to be induced solely by the presence of a live plant pathogen.
In T. hamatum, expression of the gene was induced by S. sclerotiorum, S. minor, S. cepivorum, but not by B. cinerea or by interaction with itself. A similar pattern was observed in T. atroviride and T. virens. This showed that the interaction was quite specific and not a generalised response to any fungus encountered. The three plant pathogen species, which did induce the gene, have in common that they are soilborne pathogens, whereas B. cinerea is a foliar pathogen. The three Trichoderma strains used are all effective biocontrol agents for one or more of the four plant pathogens listed above (McLean and Stewart 2000; Rabeendran 2000; McLean et al. 2005; Stewart et al. 2005; Rabeendran et al. 2006), although not all combinations have been tested; so, it is not possible to correlate biocontrol efficacy with expression of the monooxygenase gene. The T. atroviride strain LU132 was effective at controlling disease caused by B. cinerea (Stewart et al. 2005) and S. cepivorum (McLean et al. 2005) and has been developed commercially for these purposes. However, the expression of the T. atroviride monooxygenase gene in response to S. cepivorum and B. cinerea was quite different, suggesting that the mechanisms used in biocontrol may be variable depending on the pathogen species involved.
Analysis of the promoter sequence of the T. hamatum monooxygenase gene revealed numerous possible regulatory motifs, but only a few of these were in conserved positions relative to the T. atroviride promoter. One that was conserved was the PacC motif associated with gene regulation in response to pH (Denison 2000). The assays using promoter fusion mutants showed that expression of the monooxygenase gene was not stimulated by changes in pH in the absence of S. sclerotiorum. However, during interaction with S. sclerotiorum, pH had a highly significant effect on the level of expression observed. Therefore, it is likely that expression of this gene is regulated via the PacC system. The monooxygenase gene promoter also had a conserved GATA (nitrogen catabolite repression) motif, and the expression of the gene was affected by the concentration of primary nitrogen, which suggested that this gene was subject to nitrogen catabolite repression. Other Trichoderma genes associated with biocontrol, such as prb1 and chit42, were also regulated through nitrogen catabolite repression (Donzelli et al. 2001; Olmedo-Monfil et al. 2002). In contrast, expression of the monooxygenase gene did not appear to be affected by glucose concentration, suggesting that the CreA (carbon repression) motif, which was present in a conserved region of the promoter, was not functional.
The T. hamatum promoter did not show much similarity to the prb1 or chit42 promoters (Steyaert et al. 2004), and only included one of the five mycoparasitism-related (MYC) elements previously described in these genes (Cortes et al. 1998; Steyaert et al. 2003), which suggested that it was regulated independently of these other biocontrol genes. The G3 promoter also showed little similarity to that of the T. reesei homologue. This is consistent with T. reesei not being a biocontrol species and the homologous gene having a somewhat different role. However, the similarities between the T. hamatum and T. atroviride promoters, which included identical sequences of up to 30 bp, suggested the presence of additional, previously undescribed regulatory elements.
The gene knockout mutants grew normally on PDA, which is consistent with the gene not being expressed during normal growth, but being expressed specifically during antagonistic activity against other fungi. The knockout mutants were less aggressive than the wild-type in confrontation with S. sclerotiorum, which was also consistent with the gene being expressed during antagonistic activity. The gene disruption conferred a loss of biocontrol ability, significant in only one of the two mutants tested, but did not abolish biocontrol completely. Therefore, although the gene has a role in biocontrol, it is not essential. As biocontrol involves a variety of cellular activities (Kubicek et al. 2001), it is likely that numerous genes are involved; therefore, many of these genes will be making a relatively minor contribution to the biocontrol outcome such that their contribution may be difficult to detect. Additionally, some genes involved in biocontrol may have redundant roles that can be totally or partially done by other enzymes. Interestingly, knockouts of the mycoparasitism-related ech42 gene in T. atroviride did not cause loss of mycoparasitic or biocontrol activity (Carsolio et al. 1999), yet the gene is accepted as being important in biocontrol. Also, disruption of tvsp1, the prb1 homologue in T. virens, produced no significant differences from the wild-type in three of four biocontrol-related parameters (Pozo et al. 2004). This inconsistency may be associated with the inherent variability of assays involving two or three live organisms.
The confrontation and biocontrol assays were conducted using 2–3 knockout mutants, rather than just one, to confirm that the phenotypic changes observed were due to the gene knockout and not due to additional random mutations. The knockout mutants behaved similarly in these assays although there was variation in the extent to which the gene knockout impaired their ability to inhibit the growth and sclerotial production of S. sclerotiorum. This variation could be explained by non-target mutation in the knockout strains. Additional Southern blot and PCR analysis of the disrupted region of the knockout genomes were performed, but revealed no differences between them (data not shown). For further characterisation of the role of this gene in biocontrol, it would be interesting to see if overexpression of the gene resulted in an increase in the biocontrol ability of the fungus.
The T. hamatum monooxygenase gene described herein had an expression pattern consistent with a role in antagonistic activity against specific fungal plant pathogens and its disruption had a negative impact on biocontrol ability. The specificity of the gene expression was quite striking in that the gene showed no expression whatsoever except in the presence of particular plant pathogens. Its role in the antagonistic interaction and its substrate remain to be determined; however, the characteristics described here indicate its involvement in biocontrol by Trichoderma species.
References
Bosch R, Garcia-Valdes E, Moore ERB (2000) Complete nucleotide sequence and evolutionary significance of a chromosomally encoded naphthalene-degradation lower pathway from Pseudomonas stutzeri AN10. Gene 245:65–74
Carpenter M, Stewart A, Ridgway HJ (2005) Identification of novel Trichoderma hamatum genes expressed during mycoparasitism using subtractive hybridisation. FEMS Microbiol Lett 251:105–112
Carsolio C et al (1999) Role of the Trichoderma harzianum endochitinase gene, ech42, in mycoparasitism. Appl Environ Microbiol 65:929–935
Carsolio C, Gutierrez A, Jimenez B, Vanmontagu M, Herrera-Estrella A (1994) Characterization of ech-42, a Trichoderma harzianum endochitinase gene expressed during mycoparasitism. Proc Natl Acad Sci USA 91:10903–10907
Cortes C, Gutierrez A, Olmedo V, Inbar J, Chet I, Herrera Estrella A (1998) The expression of genes involved in parasitism by Trichoderma harzianum is triggered by a diffusible factor. Mol Genet Genomics 260:218–225
Denison SH (2000) pH regulation of gene expression in fungi. Fungal Genet Biol 29:61–71
Donzelli BGG, Lorito M, Scala F, Harman GE (2001) Cloning, sequence and structure of a gene encoding an antifungal glucan 1,3-β-glucosidase from Trichoderma atroviride (T. harzianum). Gene 277:199–208
Eppink MHM, Schreuder HA, Van Berkel WJH (1997) Identification of a novel conserved sequence motif in flavoprotein hydroxylases with a putative dual function in FAD/NAD(P)H binding. Protein Sci 6:2454–2458
Flores A, Chet I, Herrera Estrella A (1997) Improved biocontrol activity of Trichoderma harzianum by over-expression of the proteinase-encoding gene prb1. Curr Genet 31:30–37
Geremia RA et al (1993) Molecular characterization of the proteinase encoding gene, prb1, related to mycoparasitism by Trichoderma harzianum. Mol Microbiol 8:603–613
Hanson LE, Howell CR (2004) Elicitors of plant defense responses from biocontrol strains of Trichoderma virens. Phytopathology 94:171–176
Harman GE, Chet I, Baker R (1980) Trichoderma hamatum effects on seed and seedling disease induced in radish and pea by Pythium spp. or Rhizoctonia solani. Phytopathology 70:1167–1172
Harman GE, Howell CR, Viterbo A, Chet I, Lorito M (2004) Trichoderma species—opportunistic, avirulent plant symbionts. Nat Rev Microbiol 2:43–56
Hjeljord L, Tronsmo A (1998) Trichoderma and Gliocladium in biological control: an overview. In: Harman GE, Kubicek CP (eds) Trichoderma and Gliocladium. Taylor and Francis, London, pp 131–151
Horst LE, Locke J, Krause CR, McMahon RW, Madden LV, Hoitink HAJ (2005) Suppression of botrytis blight of begonia by Trichoderma hamatum 382 in peat and compost-amended potting mixes. Plant Dis 89:1195–1200
Howell CR (1998) The role of antibiosis in biocontrol. In: Harman GE, Kubicek CP (eds) Trichoderma and Gliocladium. Taylor and Francis, London, pp 173–184
Ishiyama D, Vujaklija D, Davies J (2004) Novel pathway of salicylate degradation by Streptomyces sp. strain WA46. Appl Environ Microbiol 70:1297–1306
Kubicek CP, Mach RL, Peterbauer CK, Lorito M (2001) Trichoderma: from genes to biocontrol. J Plant Pathol 83:11–23
Limon MC, Pintor-Toro JA, Benitez T (1999) Increased antifungal activity of Trichoderma harzianum transformants that overexpress a 33-kDa chitinase. Phytopathology 89:254–261
Liu DQ, Liu H, Gao XL, Leak DJ, Zhou NY (2005) Arg169 is essential for catalytic activity of 3-hydroxybenzoate 6-hydroxylase from Klebsiella pneumoniae M5a1. Microbiol Res 160:53–59
Lorang JM et al (2001) Green fluorescent protein is lighting up fungal biology. Appl Environ Microbiol 67:1987–1994
Lorito M, Farkas V, Rebuffet S, Bodo B, Kubicek CP (1996) Cell wall synthesis is a major target of mycoparasitic antagonism by Trichoderma harzianum. J Bacteriol 178:6382–6385
Lutz MP, Wenger S, Maurhofer M, Defago G, Duffy B (2004) Signaling between bacterial and fungal biocontrol agents in a strain mixture. FEMS Microbiol Ecol 48:447–455
McLean KL, Stewart A (2000) Application strategies for control of onion white rot by fungal antagonists. N Z J Crop Hortic Sci 28:115–122
McLean KL, Swaminathan J, Frampton CM, Hunt JS, Ridgway HJ, Stewart A (2005) Effect of formulation on the rhizosphere competence and biocontrol ability of Trichoderma atroviride C52. Plant Pathol 54:212–218
McPherson MJ (2000) PCR. BIOS Scientific Publishers, Oxford
Meyer GD, Bigirimana J, Elad Y, Hofte M (1998) Induced resistance in Trichoderma harzianum T39 biocontrol of Botrytis cinerea. Eur J Plant Pathol 104:279–286
Migheli Q, Gonzalez Candelas L, Dealessi L, Camponogara A, Ramon-Vidal D (1998) Transformants of Trichoderma longibrachiatum overexpressing the β-1,4-endoglucanase gene egl1 show enhanced biocontrol of Pythium ultimum on cucumber. Phytopathology 88:673–677
Olmedo-Monfil V, Mendoza-Mendoza A, Gomez I, Cortes C, Herrera Estrella A (2002) Multiple environmental signals determine the transcriptional activation of the mycoparasitism related gene prb1 in Trichoderma atroviride. Mol Genet Genomics 267:703–712
Pozo MJ, Baek JM, Garcia JM, Kenerley CM (2004) Functional analysis of tvsp, a serine protease-encoding gene in the biocontrol agent Trichoderma virens. Fungal Genet Biol 41:336–348
Punt PJ, Oliver RP, Dingemanse MA, Pouwels PH, van den Hondel CAMJJ (1987) Transformation of Aspergillus based on the hygromycin B resistance marker from Escherichia coli. Gene 56:117–124
Rabeendran N, Biological control of Sclerotinia diseases of vegetables, Ph.D. Thesis, Lincoln University, Canterbury, New Zealand, 2000
Rabeendran N, Jones EE, Moot DJ, Stewart A (2006) Biocontrol of Sclerotinia lettuce drop by Coniothyrium minitans and Trichoderma hamatum. Biol Control 39:352–362
Rocha-Ramirez V, Omero C, Chet I, Horwitz BA, Herrera Estrella A (2002) Trichoderma atroviride G-protein α-subunit gene tga1 is involved in mycoparasitic coiling and conidiation. Eukaryot Cell 1:594–605
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Shoresh M, Yedidia I, Chet I (2004) Involvement of the jasmonic acid/ethylene signaling pathway in the systemic resistance induced in cucumber by Trichoderma asperellum T203. Phytopathology 95:76–84
Stewart A et al (2005) Biological control of Botrytis diseases by the antagonistic fungus Trichoderma atroviride. In: Proceedings of the 15th Austalasian Plant Pathology Society, Geelong, Australia, p 83
Steyaert JM, Ridgway HJ, Elad Y, Stewart A (2003) Genetic basis of mycoparasitism: a mechanism of biological control by species of Trichoderma. N Z J Crop Hortic Sci 31:281–291
Steyaert JM, Stewart A, Jaspers MV, Carpenter M, Ridgway HJ (2004) Co-expression of two genes, a chitinase (chit42) and proteinase (prb1), implicated in mycoparasitism by Trichoderma hamatum. Mycologia 96:1245–1252
Tsuji H, Oka T, Kimoto M, Hong Y-M, Natori Y, Ogawa T (1996) Cloning and sequencing of cDNA encoding 4-amino benzoate hydroxylase from Agaricus bisporus. Biochim Biophys Acta 1309:31–36
Tunnacliffe A, van de Vrugt H, Pensotti V, Radice P (1996) The coatomer protein δ-COP, encoded by the archain gene, is conserved across diverse eukaryotes. Mamm Genome 7:784–786
Vallon O (2000) New sequence motifs in flavoproteins: evidence for common ancestry and tools to predict structure. Proteins 38:95–114
Vasseur V, Montagu Mv, Goldman GH (1995) Trichoderma harzianum genes induced during growth on Rhizoctonia solani cell walls. Microbiology 141:767–774
Zeilinger S (2004) Gene disruption in Trichoderma atroviride via Agrobacterium-mediated transformation. Curr Genet 45:54–60
Zeilinger S et al (1999) Chitinase gene expression during mycoparasitic interaction of Trichoderma harzianum with its host. Fungal Genet Biol 26:131–140
Zeilinger S, Reithner B, Scala F, Peissl I, Lorito M, Mach RL (2005) Signal transduction by Tga3, a novel G protein α subunit of Trichoderma atroviride. Appl Environ Microbiol 71:1591–1597
Zhang S, Monahan BJ, Tkacz JS, Scott B (2004) Indole-diterpene gene cluster from Aspergillus flavus. Appl Environ Microbiol 70:6875–6883
Acknowledgments
This work was funded by the New Zealand Foundation for Research, Science and Technology, contract LINX0201. pYT6 was kindly provided by Professor Barry Scott of Massey University, New Zealand. pCT74 was kindly provided by Lynda Ciuffetti of Oregon State University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by G. Braus.
Rights and permissions
About this article
Cite this article
Carpenter, M.A., Ridgway, H.J., Stringer, A.M. et al. Characterisation of a Trichoderma hamatum monooxygenase gene involved in antagonistic activity against fungal plant pathogens. Curr Genet 53, 193–205 (2008). https://doi.org/10.1007/s00294-007-0175-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-007-0175-5