
Abstract
A transformation system utilizing Agrobacterium tumefaciens was developed for targeted gene disruption in Monilinia fructicola, a fungal pathogen that causes brown rot disease of stone fruits. Transformation with a vector containing the neomycin phosphotransferase II (nptII) cassette flanked with 4 kb cutinase gene (Mfcut1) flanking sequences resulted in an average of 13 transformants per 105 spores. When assayed by PCR and DNA blot analyses, more than 50% of the transformants recovered had integrated in the targeted Mfcut1 locus. Both target-gene-specific and non-specific integrations carried direct (head-to-tail) repeat T-DNA integrations. Sequence analysis of these T-DNA integrations revealed that 26 bp of the T-DNA right border were missing at the junctions between direct repeats in all cases. The recombination event during non-specific T-DNA integration in this fungus was unlike that reported in Agrobacterium-mediated transformation in plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Monilinia fructicola (G. Wint.) Honey, a filamentous ascomycete, is one of the most destructive pathogens of stone fruit species worldwide. This fungus causes severe blossom blighting and pre- and post-harvest fruit rots, resulting in serious economic losses for growers and processors (Ogawa 1991). Macroconidia constitute the major form of primary and secondary inoculum, and infection of plant tissues results from the ability of the fungus to penetrate directly through the cuticle or through natural openings and wounds on the host (Curtis 1928). However, relatively little is known about the molecular basis for pathogenicity of this fungus, due, in part, to the lack of an efficient transformation system as well as the complications imposed by the multi-nucleate nature of M. fructicola cells, thus making it difficult to create stable mutants.
Targeted gene disruption is a valuable method for analysis of gene function in filamentous fungi. Homologous gene replacement can precisely disrupt a target gene by replacing a portion of that gene with a selectable marker. This disruption is brought about by homologous recombination between the host gene and the introduced DNA. In fungi, homologous recombination efficiency varies depending on the species and the length of homologous regions used (Bird and Bradshaw 1997; Gray and Honigberg 2001). In Saccharomyces cerevisiae, homologous gene replacement occurred with high efficiency (as many as 75% of the transformants) with a 40 bp homologous region (Longtine et al. 1998). Filamentous fungi have a lower homologous recombination rate and require a longer flanking region to improve the frequency. In Neurospora crassa, a 30% homologous recombination rate was obtained using homologous flanking sequences of 9 kb (Asch and Kinsey 1990). In Botrytis cinerea, a member of the same family as M. fructicola, a 25% homologous recombination rate was obtained using 2 kb of a homologous sequence (van Kan et al. 1997).
A functional understanding of pathogenicity and development in M. fructicola will require an efficient transformation system and effective methods for targeted gene disruption. Currently, the two most common methods for introducing DNA into filamentous fungi are protoplast transformation utilizing CaCl2/PEG, and Agrobacterium tumefaciens-mediated transformation. CaCl2/PEG transformation has been used successfully with many phytopathogenic fungi. Recently, Agrobacterium-mediated transformation has been used for transformation of fungi of importance in industry and agriculture (Michielse et al. 2005b). With this transformation method, homologous gene replacement has been reported in species of Aspergillus, Trichoderma, Mycosphaerella, Verticillium, Fusarium, Leptosphaeria and Magnaporthe. When compared to CaCl2/PEG transformation, Agrobacterium-mediated transformation provided a three- to sixfold increase in the frequency of gene replacement in Aspergillus awamori (Michielse et al. 2005a). In Verticillium fungicola, a 75% homologous integration frequency was achieved using Agrobacterium-mediated transformation (Amey et al. 2003).
Agrobacterium tumefaciens is a plant pathogen that causes crown gall disease by transfer of T-DNA carrying tumorigenesis genes into the plant genome. The T-DNA transfer is regulated by virulence genes located on the Ti plasmid of A. tumefaciens. The expression of these genes is induced by plant phenolic compounds such as acetosyringone (Lee et al. 1995; Gelvin 2003). When acetosyringone is present during the co-cultivation of fungal cells and Agrobacterium cells, T-DNA is transferred from the bacterial cells to the fungal cells as in plants (Bundock and Hooykaas 1996). VirD2 and VirE2 are two important virulence proteins required for the delivery of the T-DNA into plant cells. VirD2 is a site-specific nuclease that nicks at the right and left borders of the T-DNA. These borders are comprised of 24–26 bp direct repeats. The action of VirD2 results in release of a single strand of T-DNA from the Ti-plasmid. VirD2 remains covalently attached to the 5′ end and guides the T-strand, now bound with VirE2, into the nucleus of a recipient cell (Durrenberger et al. 1989; Mayerhofer et al. 1991; Bundock and Hooykaas 1996). VirD2 may be involved with the precision of the T-DNA integration (Tinland et al. 1995), but does not seem to play an essential role in the integration process. Host factors, enzymes and the repair system for double-strand breaks also have been implicated in the process of T-DNA integration in plants (Salomon and Puchta 1998). Similarly, specific and non-specific T-DNA integration appears to be regulated by host proteins in yeast (van Attikum et al. 2001).
The fate of T-DNA after its transfer to a recipient cell has not been well studied in filamentous fungi. In plants, T-DNA can integrate at random positions into the genome by non-specific integration through illegitimate recombination, even when the T-DNA carries sequences with extensive homology to specific sequences within the plant genome. In addition, T-DNA is often reported to integrate as a series of direct (head-to-tail) and/or indirect (head-to-head or tail-to-tail) repeats. In S. cerevisiae, T-DNA preferentially integrates by homologous recombination when it shares homology with yeast sequences. However, in the absence of DNA homology, integration occurs by illegitimate recombination as observed in plants (Bundock et al. 1995; Risseeuw et al. 1996). Nucleotide deletions and incorporation of filler DNA often occur at the junctions between two T-DNAs and between T-DNA and integrated genomic DNA in yeast and plants (Bundock and Hooykaas 1996; Krizkova and Hrouda 1998; Dietz-Pfeilstetter et al. 2003; Tzfira et al. 2004). Analysis of these junctions and the structures of the integrated T-DNA has provided the basis for a proposed model for T-DNA integration in plants (Krizkova and Hrouda 1998; Dietz-Pfeilstetter et al. 2003; Tzfira et al. 2004).
The Mfcut1 gene encodes a cutinase (EC 3.1.1.74) and occurs as a single copy in the genome of M. fructicola. The expression of Mfcut1 is regulated by plant host factors, and the flanking regions of the Mfcut1 gene recently have been isolated (Wang et al. 2002; M-H Lee and R.M. Bostock, unpublished). In this study, Mfcut1 was targeted to establish an Agrobacterium-mediated transformation system for gene disruption. Here, the efficiency of Agrobacterium-mediated transformation in M. fructicola is reported. This study is the first to characterize gene replacement and patterns of T-DNA integration in the genus Monilinia.
Materials and methods
Strains, plasmids and cultivation
Monilinia fructicola strain MUK-1, a single spore isolate from an infected peach fruit from a local orchard (Bostock et al. 1999), was used throughout this work. Fungal strains used for transformation and genomic DNA isolation were grown in clarified V8 (cV8) agar medium at room temperature for 1 week. A supplemented minimal medium (SMM), containing 0.07% NH4NO3, 0.12% K2HPO4, 0.036% NaH2PO4, 0.03% MgSO4, 1% glucose and 20 μl/l Vogel’s trace elements (Pratt and Aramayo 2002), was used as a basal medium for fungal transformation and maintenance of transformants. SMM containing 50 μg/ml G-418 sulfate (A.G. Scientific, Inc. USA) (SMMG50) was used to select and maintain transformants. All fungal strains were stored in 10% DMSO at −75°C for long-term storage. A. tumefaciens strain AGL.1 (C58, RecA, pTiBo542ΔT-DNA) (Hellens et al. 2000) was used for transformation experiments, and was maintained in AMM medium (Bundock et al. 1995) containing rifampicin (50 μg/ml) and carbenicillin (50 μg/ml) at 28°C. Escherichia coli strain XL1-Blue (Stratagene, USA) was used for manipulations of all plasmids. Binary vector pCAMBIA1300 (CAMBIA, Australia) was used as a backbone for constructing the gene replacement vector and its sequence is in GenBank (#AF234296). Plasmid pART27 (also called pHELLSGATE; GenBank accession no. AJ311874) was kindly provided by Dr. Linda Ciuffeti (Oregon State University) and used as a template to amplify the neomycin phosphotransferase gene (nptII) cassette conferring resistance to G-418. PCR products from the junction of T-DNA repeats were cloned into a pGEM-T Easy vector (Promega, Co. USA) for sequencing.
Construction of the Mfcut1 homologous replacement vector
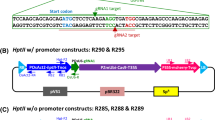
A 4.3 kb fragment containing the Mfcut1 ORF and its flanking regions (GenBank accession no. DQ173196) was amplified from M. fructicola genomic DNA by high fidelity PCR using the KOD HiFi polymerase (Novagen, USA) with primers, ScXb-F (forward) and ScXb-R (reverse) (Table 1). A SacI site was added to the forward primer and an XbaI site was added to the reverse primer. The PCR product was gel purified and cloned into SacI/XbaI-digested pCAMBIA1300 to produce plasmid pMFCU-1300. The 350 bp ApaI/BamHI fragment of the Mfcut1 coding region in pMFCU-1300 was removed and replaced with a 1.9 kb nptII cassette amplified from pART27 by primers nptII-Apa and nptII-Bgl (Table 1) using KOD HiFi polymerase. The resulting plasmid, pMFCUN-23 (Fig. 1) has the Mfcut1 replacement construct in which the nptII cassette replaces a portion of the Mfcut1 ORF with flanking regions of 2 kb of the Mfcut1 sequence on each side. pMFCUN-23 (14.5 kb) was transformed into A. tumefaciens AGL.1 and used for Agrobacterium-mediated transformation and gene disruption in M. fructicola. PCR amplification using the KOD HiFi polymerase kit was conducted according to the manufacturer’s instructions.

Map and gene replacement of Mfcut1. The replacement T-DNA contains a G418-resistance cassette (nptII) flanked by border sequences of Mfcut1. By specific integration through homologous recombination, 350 bp of the Mfcut1 coding region was replaced by the nptII cassette. LB and RB are the 26 nucleotides of left and right T-DNA borders, respectively. Primers for PCR analysis are indicated by black arrows, and hollow arrows indicate the direction of transcription of Mfcut1 and the two antibiotic resistance cassettes. The size of each fragment is indicated
Fungal transformation
Agrobacterium tumefaciens AGL.1 carrying pMFCUN-23 was grown in LB containing kanamycin at 28°C overnight and then transferred into the transformation-inducing broth containing 200 μM acetosyringone for an additional 4–6 h at 28°C (Bundock et al. 1995). A spore suspension of M. fructicola was freshly prepared from cultures growing on cV8 agar medium by flooding the plate with sterile deionized water, and then filtering the suspension through miracloth (Calbiochem, USA). The spore concentration of the filtrate was determined with a hemocytometer and the filtrate was diluted in water to achieve the indicated concentration.
The transformation procedure followed published protocols (Bundock et al. 1995; Covert et al. 2001) with modifications. A 100 μl spore suspension was mixed with a 100 μl pre-induced bacterial culture with an optical density of 0.4 at 660 nm and then spread on a 0.45 μm nitrocellulose membrane (Whatman, USA). The membranes were placed on an inducing-medium agar plate (Bundock et al. 1995) and incubated at 19–21°C for 2 or 3 days. At least ten inducing plates were inoculated in this manner for each transformation experiment. After incubation, membranes were placed either top-side-up or top-side-down on SMM selective agar medium containing 50 μg/ml G-418, 50 μg/ml cephalexin, 25 μg/ml vancomycin, and 50 μg/ml streptomycin (SMMG50cvs). Each membrane was removed after 7 days of incubation at room temperature, and hyphal tips from individual fungal colonies that grew into the agar medium were transferred to fresh SMMG50 agar plates.
Genomic DNA isolation and PCR amplification
Fungal mycelium (20–50 mg) was frozen in liquid nitrogen in a 1.5 ml tube (Fisher Scientific), ground into powder using a chilled pellet pestle (Nalgene), extracted with 300 μl lysing buffer (50 mM Tris pH 8.0, 50 mM EDTA, 3% SDS and 0.1% 2-mercaptoethanol) and incubated at 65°C for 1 h. The lysing mixture then was centrifuged and the supernatant was collected for DNA extraction. The supernatant was extracted with an equal volume of phenol:chloroform (1:1) and precipitated with isopropanol and sodium acetate. The precipitate was collected by centrifugation, and the pellet was washed with 70% ethanol. DNA pellets were dissolved in 50 μl TE buffer containing RNase (20 μg/ml) and incubated at 4°C overnight. Two microliters of the genomic DNA solution were diluted to 100 μl with TE buffer for PCR to minimize inhibitors present in the original solution. Alternatively, genomic DNA was isolated with the MasterPure Plant leaf DNA purification kit (Epicentre Biotechnologies, USA) according to the manufacturer’s instructions, and was used without any dilution for PCR reaction using the KOD HiFi polymerase kit. PCR primers nptII-Apa and nptII-Bgl were used to amplify the nptII cassette, primers 4.3 mf and 23–2,030 were used to examine the 5′ end replacement, and primers ‘left-region’ and ‘right-region’ were used to examine the junctions of T-DNA repeats (Fig. 1). Taq polymerase (Promega, Co. USA) was used for routine PCR amplification unless otherwise indicated. The sequences of primers used in this experiment are listed in Table 1. Amplification of the 5′-end replacement fragment was performed with Taq polymerase for 40 cycles to increase DNA yields. PCR products were visualized by electrophoresis using 0.8% or 1.0% agarose gels that were stained with ethidium bromide.
Southern blot analysis
One microgram of genomic DNA was digested with restriction enzymes overnight. The concentration of digested DNA was quantified by fluorometry with Hoechst 33258. Five hundred nanograms of digested DNA was separated on a 0.7% agarose gel and transferred overnight to a Hybond-N+ membrane (Amersham Pharmacia Biotech, USA). The AlkPHOs Direct Labeling kit (Amersham Pharmacia Biotech) was used for probe labeling and hybridization. Each of the three probes used in this procedure is approximately 500 bp in length. Probes were PCR-amplified by Taq polymerase from pMFCUN-23 and gel purified after electrophoresis. Probe labeling with alkaline phosphatase and DNA hybridizations were performed according to the manufacturer’s instructions (Amersham Pharmacia Biotech, USA). Hybridizations were performed overnight at 55°C and primary washes were performed at 65°C. The probes were detected with CDP-Star chemiluminescent substrate (Sigma-Aldrich, USA) for alkaline phosphatase.
Sequence analysis
DNA sequence analysis with the ABI Prism Bigdye Terminator sequencing reaction mix (Applied Biosystems, Foster City, CA, USA) was performed in both directions at the UC Davis DNA Sequencing Facility. Sequence alignments were conducted using Vector NTI AdvanceTM 9 (Invitrogen, Co).
Nuclear staining
Monilinia fructicola MUK1 was grown in SMM, cV8 and YPD (0.1% yeast extract, 0.1% peptone, 0.1% glucose) media at room temperature for 2 days. Fungal hyphae were stained with 2 μg/ml DAPI (4′,6-diamidino-2-phenylindole) for 1 min and examined under a Nikon fluorescence microscope.
Results
Determination of nuclei in M. fructicola
To assess the numbers of nuclei in M. fructicola, the wild-type MUK1 isolate was grown on different media (Fig. 2). MUK1 contained more than eight nuclei in a single hyphal cell when grown in rich complete cV8 and weak complete YPD medium. When grown in the minimal medium (SMM), three nuclei typically were observed in hyphal cells. Thus, transformants grown in SMM were used for hyphal tip isolations.

Nuclei in hyphal cells of M. fructicola MUK1 grown in SMM, cV8 or YPD medium. Growing hyphae were stained with DAPI and visualized by fluorescence microscopy to determine the number of nuclei in cells. Arrows indicate the position of septa delimiting representative hyphal cells. Upper panels are photomicrographs of hyphae viewed under visible light. Lower panels are the same fields but stained with DAPI and viewed under UV light. Bars 20 μm
Transformation efficiency
Fungal colonies that survived on the surface of the co-cultivation membranes after 7 days of incubation on selective medium (SMMG50) were presumed to be transformants. Putative M. fructicola transformants not only could grow in and below the surface of selective agar, but also could support growth of aerial hyphae. Pseudo-transformants grew on the surface but did not produce aerial hyphae under the same conditions. After four cycles of hyphal tip purification, a putative transformant was considered a true transformant when it produced aerial hyphae on SMMG50. Among all experiments, 50% of the putative transformants isolated from the first selective plates and then passed through four cycles of hyphal tip purification were stably resistant to G-418 sulfate.
To recover all transformants in a short incubation period before most colonies merged, co-cultivation membranes were placed on selective media with the top-side down instead of the top-side up. In experiments 3 and 5, the membranes with the top-side-down had twice the number of transformants as those with the top-side-up (Table 2). Increasing the co-cultivation period on inducing medium from 2 to 3 days did not appear to increase the transformation rate. Doubling the number of spores during transformation increased the transformation rate. No transformants were obtained when transformations were conducted with either A. tumefaciens AGL-1 (without binary vector) or the A. tumefaciens AGL-1 carrying pCAMBIA1300 (no Mfcut1 replacement construct). Also no transformants were obtained when acetosyringone was absent during co-cultivation. The average transformation rate over five experiments was 13 transformants per 105 spores.
PCR screening for T-DNA transfer
Homologous gene replacement requires that the replacement construct adjacent to the right border of the T-DNA is transferred into the fungal cell (Fig. 1). Since T-DNA transfer begins from the right border, PCR was used to screen for the presence of the Mfcut1 5′-end flanking region adjacent to the nptII cassette. The ScXb-F and nptII-Bgl primers were used to amplify a 3.7-kb fragment containing the fused Mfcut1 and nptII cassette. In total, 109 of the 115 transformants from five experiments yielded the 3.7 kb fragment (Fig. 3), indicating that the construct was transferred successfully and integrated into the genome of M. fructicola. The remaining six transformants with no detectable 3.7 kb PCR fragment could be either pseudo-transformants or may carry truncated T-DNA with the nptII cassette. To confirm the latter, PCR was performed to detect the entire nptII cassette using the nptII-Apa and nptII-Bgl primers. The 1.9 kb PCR amplification product containing the full-length nptII cassette was obtained from all six transformants (data not shown), suggesting there was truncated T-DNA transfer in these six transformants. No PCR product was amplified from wild-type DNA under the same conditions using the two sets of primers. All transformants retained resistance to G418 sulfate after several sequential cultures on non-selective medium. Thus NptII appeared to be a stable selectable marker for M. fructicola.

PCR analysis of genomic DNA with primers (ScXb-F and nptII-Bgl) specific for the amplification of the 5′ end fragment of the Mfcut1 replacement construct (3.7 kb). The sizes of standard DNA are indicated. The gel illustrates representative results (36 transformants) from a total of 115 true transformants analyzed by PCR. A transformed strain (T in upper gel) and the wild-type strain (WT in lower gel) do not carry the 3.7 kb PCR product. The other 35 transformants all have the 3.7 kb PCR fragments. The 3.7 kb fragment is also amplified from the pMFCUN-23 vector (V in lower gel). The map of the replacement T-DNA and the corresponding locations of the two primers are shown at the top of the figure
Analysis of homologous gene replacement
Initially, we attempted to determine replacement of the Mfcut1 gene by using the cut-F2 and cut-R2 primers that specifically recognize the Mfcut1 ORF. The primers were designed to amplify a DNA fragment from the wild-type strain, but would not amplify any product from a transformant if homologous disruption occurred at Mfcut1. However, a 350 bp fragment was amplified from all 115 transformants screened (data not shown). Since M. fructicola cells are multinucleate, even if one nucleus within a cell carries the normal gene, Mfcut1 will be detected by using these two primers. We then designed and used primers that would specifically amplify a homologous replacement of Mfcut1. The primer 4.3 mf, specific to the upstream region of the Mfcut1 ORF but not present on pMFCUN-23, and the primer 23–2,030, specific to the NOS terminator fused to nptII in pMFCUN-23, were used for this purpose (Fig. 4). If transformants contained the replacement Mfcut1 fragment in the M. fructicola genome, a 2 kb fragment containing the 5′ end Mfcut1 fused with the nptII cassette would be amplified by primers 4.3 mf and 23–2,030. No 2 kb fragment amplification would be expected from non-specific integrations. Analysis of the PCR products by gel electrophoresis indicated that 12% of 115 transformants carried a 5′ end replacement fragment of the correct size. The presence of the 2-kb DNA fragment from the PCR indicates that specific replacement occurred within the Mfcut1 gene in some transformants.

PCR analyses for the 5′ end replacement of Mfcut1 genomic DNA with undiluted genomic DNA by KOD HiFi polymerase. Transformants were analyzed with primers 4.3 mf and 23–2,030 specific for amplification of the 5’ end of the replaced fragment (2 kb). Lane 1 contains 1 kb and standard DNA ladders (Gibco BRL). Lanes 2–9 correspond to transformants H38, L21, L40, I78, M23, M27, M36 and M249, respectively. All transformants carry the 2 kb product except M27 (lane 7)
It is likely that transformants with only one of their nuclei carrying the replacement fragment might escape detection by gel electrophoresis of the PCR amplified products. Therefore, DNA gel blot hybridization was used to better visualize the PCR products. Hybridization with a DNA probe (probe 1) that included part of the nptII cassette sequence and Mfcut1 flanking regions revealed that 52% of the 115 analyzed transformants carried the 2 kb fragment.
These results indicate that some transformants harbor the disrupted Mfcut1 allele in some, but not all, of their nuclei. Several transformants with no detectable 2 kb fragment in ethidium-stained gels presented a 2 kb band when hybridized with the probe, confirming low levels of the disrupted Mfcut1 allele in total DNA from these transformants. In addition, fragments larger or smaller than 2 kb were amplified. These may be products from multiple non-specific integrations that are amplified under conditions used for this PCR analysis (40 cycles using a Taq polymerase without error-proof function).
We also designed another set of primers specific to the 3′ end replacement fragment. However, a non-specific fragment similar in size to that expected for the target also was amplified. Adjustment of the PCR conditions did not eliminate the non-specific fragment from the product mix. A PCR reaction with KOD HiFi polymerase using the primers 4.3 mf and 23–2,030 was performed to amplify the 2 kb replaced fragment on the 5′ end. The PCR amplification program listed in Table 1 was the same as that used to achieve the results above except that undiluted genomic DNA purified with the Epicentre DNA purification kit was used as template. Eight transformants were analyzed in this way. As shown in Fig. 4, all examined transformants carried the 2 kb product except M27. The 2 kb PCR products from H38 and L40 were purified and sequenced, which confirmed that they represent the Mfcut1 replacement fragment at the 5′ end (Fig. 5). These results indicate the presence of the disrupted Mfcut1 locus in some nuclei.

Partial sequence alignments of the 2 kb 5′ end replaced fragments with the predicted Mfcut1 disrupted genomic locus. PCR products of the 2 kb replaced fragments from H38 and L40 (Fig. 4) were purified and sequenced with primers 4.3 mf and 23–2,030 (boxes). a Sequences of H38-4.3 mf and L40–4.3 mf were obtained with primer 4.3 mf for the 5′ end of the 2 kb products. b Sequences of H38-23–2,030 and L40-23–2,030 were obtained with primer 23–2,030 for the 3′ end of the 2 kb products. Sequencing orientations are indicated by the arrows. Sequences in front of the straight vertical line and delimited by the black solid line are present only in the M. fructicola Mfcut1 locus, whereas sequences after the vertical line and delimited by the dotted line are present in the Mfcut1 homologous replacement construct of pMFCUN-23. Numbers in parentheses indicate the number of nucleotides in that sequence
As a strategy to obtain transformants with homologous nuclei harboring the disrupted Mfcut1, 23 transformants carrying only the single 2 kb product were chosen for further purification by four sequential cycles of single hyphal tip transfer on SMMG50.
Integration of T-DNA into the genome of M. fructicola
To examine the T-DNA integration patterns and to confirm the Mfcut1 disruption in M. fructicola, the 23 transformants and three others (M17, M27 and M28) that did not carry the 5′ end Mfcut1 replaced fragment, were subjected to Southern hybridization analysis. Genomic DNA was digested with BamHI, and hybridized with Probe 1. As shown in Fig. 6, all analyzed transformants carried more than one copy of the T-DNA, suggestive of non-specific integration. All transformants retained the wild-type 2.2 kb Mfcut1 sequence and some also carried the 3.5 kb Mfcut1 replacement fragment. Most transformants carried the 8.5 kb fragment corresponding to the full-length T-DNA. The intensity of this band was much higher than that of the 2.2 kb Mfcut1 sequence, suggesting that T-DNA might integrate into the genome as a series of direct repeats. Truncated T-DNA integration was found in strain M254, in which the fragment size of the integrated DNA is smaller than the 3.5 kb Mfcut1 replaced fragment, and the density of this band is similar to the 2.2 kb Mfcut1 fragment (hollow arrows in Fig. 6).

Southern hybridization of specific and non-specific T-DNA integrations. Genomic DNA was digested with BamHI and hybridized with probe 1. The wild-type strain (WT) and 25 transformants after four rounds of single hyphal tip purification were used in this analysis. The hybridized fragments of Mfcut1 (2.2 kb), Mfcut1 replaced (3.5 kb) and T-DNA direct repeats (8.5 kb) are indicated by black arrows. The truncated T-DNA is indicated by a hollow arrow. Some of the faint 3.5 kb replaced fragments are shown by solid arrow heads. The hollow arrowheads indicate approximately 11.8 kb fragments which might be generated by tail-to-tail indirect repeat integrations or by multiple single T-DNA integration in the same locus. The map of the nptII-replaced Mfcut1 locus in genomic DNA and the corresponding location of probe 1 used in this experiment are illustrated at the top of the figure
Some of the transformants, such as M249 and M265, displayed only faint 3.5 kb bands for the replacement sequence (indicated with arrowheads in Fig. 6) and some of the transformants such as M27 did not have a 3.5 kb hybridizing band despite being positive for Mfcut1 replacement from previous PCR screening. The intensity of the hybridizing 3.5 kb band in these transformants could arise from the low ratio of the numbers of nuclei containing the Mfcut1 replacement to the numbers of nuclei containing wild-type Mfcut1. Serial hyphal-tip isolation on the selective minimal medium failed to obtain any homogeneous Mfcut1disruptants.
Analysis of the junctions of T-DNA repeats
To determine if the T-DNA was integrated as repeats in the genome, the T-DNA junctions in the 26 transformants were examined by PCR (25 from Fig. 6 and L39 from Fig. 8). Most transformants analyzed had PCR products between 400 and 600 bp when the right region-specific and left region-specific primers were used together (Fig. 7). The predicted PCR products of precise direct and indirect T-DNA repeats with no deletions or filler nucleotides would be 600 bp for head-to-tail direct repeats, 582 bp for head-to-head indirect repeats, and 618 bp for tail-to-tail indirect repeats. M273 is the only exception where a 2-kb fragment was amplified (Fig. 7), which is likely due to the integration of two T-DNAs in the same locus with the same orientation. Some transformants such as M36 produced more than one small PCR product, possibly resulting from the coexistence of multiple copies of direct and indirect repeats or by non-specific amplification. However, none of the transformants yielded a PCR product smaller than 2 kb that was apparent in agarose gels when either the right region-specific (for the junction of head-to-head repeats) or left region-specific primer (for the junction of tail-to-tail repeats) was used individually to drive the reaction (data not shown). Some transformants (H38, L21, M17, M249, and M314) showed a strong fragment of approximately 11.8 kb, which is close in size to a tail-to-tail indirect repeat (Fig. 6, Table 3). However, there is no PCR product smaller than 2 kb detectable by gel electrophoresis arising from the junction between indirect repeats. A faint 3 kb band was detected under the same conditions with M17 and M249 (data not shown). We cannot rule out the possibility of T-DNA integration as head-to-head indirect repeats because this orientation is not detectable by probe 1 when hybridized to BamHI digested genomic DNA (Fig. 6, Table 3).

PCR analysis of genomic DNA from 26 transformants in Figs. 4 and 6. Primers specific for the amplification of the junction of T-DNA repeats were used. The sizes of DNA standards are indicated. All transformants except M273 yielded small fragments (400–650 bp) similar to the predicted sizes for fragments between junctions. A 2 kb fragment was amplified from M273 (arrow). No PCR product was detected in the wild-type (WT) and pMFCUN-23 (V) strains. The DNA standard markers are indicated (MK). The map of the replacement T-DNA and the corresponding locations of the two primers are shown at the top of the figure
To further assess the orientation of the T-DNA repeats, six transformants (H61, L39, I78, M35, M254 and M307) yielding simple integration patterns in previous Southern hybridization analyses were analyzed using the hptII or nptII probe to hybridize to EcoRI or SacI digested genomic DNA (Fig. 8). The predicted sizes of DNA fragments after the hybridization are listed in Table 3. All six transformants had 8 kb EcoRI and 8.5 kb SacI hybridizing bands when hybridized with an hptII or nptII probe, indicating that the six transformants carry the head-to-tail direct repeat integration. Some nuclei in M307 appear to carry a tail-to-tail indirect repeat integration as assessed with the hptII probe (arrows in Fig. 8). The head-to-head indirect repeat integration was detected in L39 by using the nptII probe (hollow arrows in Fig. 8). However, the fragment densities of the two types of indirect repeats are much lower than the direct repeat fragments, suggesting the total copy numbers of T-DNA in the indirect repeats may be lower than that of the direct repeats. This could explain why indirect repeats are undetectable by PCR when using either the right region-specific (for the junction of head-to-head repeats) or left region-specific primer (for the junction of tail-to-tail repeats) alone.

Southern hybridization of direct and indirect repeat T-DNA integrations of six transformants. Genomic DNA was digested with either EcoRI (E) or SacI (S) and then hybridized with probes corresponding to hptII or nptII, as described in the Materials and methods. The resulting direct repeat fragments (approximately 8 and 8.5 kb) are marked by rectangles. The corresponding fragments of tail-to-tail indirect repeats in M307 after the hptII hybridization are indicated by arrows. The corresponding fragments of head-to-head indirect repeats in L39 after the nptII hybridization are indicated by hollow arrows. See Table 3 for predicted fragment sizes of direct and indirect repeats. The map of the replacement T-DNA and the corresponding locations of the two primers are shown at the top of the figure
To determine if the integration of T-DNA resulted in deletion or insertion, PCR products of direct repeat junctions were sequenced and analyzed. Seven transformants (H61, I78, L39, M35, M254, M307 and M177) recovered from seven co-cultivation plates in two transformation experiments were used in this analysis. Sequence analysis of the amplified products indicated that all these transformants except M35 contained no extra nucleotides (filler) at the junctions (Fig. 9). In contrast, the transformant M35 contained an imprecise junction with a five-nucleotide filler. A 26 bp sequence in the right border and 4–28 bp of adjoining nucleotides of T-DNA were deleted during integration in all transformants analyzed. In addition, 9–15 nucleotides at the left borders in all transformants were also missing. No deletion was detected in the T-DNA adjoining region at the left border in any transformant except M177 (Table 4, Fig. 9).

Junctions of direct repeat integrated T-DNAs. The sequences of each border are indicated along with the adjoining T-DNA sequence of pMFCUN-23. The 26 bp of each border sequence are underlined. The residual sequences of the seven transformants are presented and are aligned to the original sequences of border regions in pMFCUN-23. The numbers indicate the number of deleted nucleotides. The filler is shown in lower case
Discussion
We have developed an efficient Agrobacterium-mediated transformation system using neomycin resistance as a selectable marker that is stable within M. fructicola. We also provide the first evidence of homologous gene replacement in this species, using a cutinase-encoding gene as a target. Evidence for high frequency direct repeat integration of T-DNA in fungi is described in detail. During the course of this study, another group used Agrobacterium-mediated transformation to generate a hygromycin-resistant mutant of M. fructicola (Dai et al. 2003). However, the transformation efficiency reported was low (one transformant per 108 conidia), and no information about T-DNA integration was provided.
The results of our transformation experiments demonstrate that M. fructicola, similar to other fungi, is highly receptive to Agrobacterium-mediated T-DNA transfer. The concentration and growth rate of bacterial cells during co-cultivation with fungal spores was found to be important for efficient transformation. Unlike other systems (Covert et al. 2001; Meyer et al. 2003), cellophane or black filter paper did not work well for co-cultivating M. fructicola and Agrobacterium. At high spore densities (i.e., >2×105 spores/ml), the number of pseudo-transformants increased. The length of the co-cultivation period is important in other fungi and has varying effects on both transformation efficiency and T-DNA integration (Meyer et al. 2003; Leclerque et al. 2004). In M. fructicola, 2-day co-cultivation is sufficient for T-DNA transfer and integration. Transformants of M. fructicola did not grow well on the top surface of the nitrocellulose membrane on selective medium. However, this did not present a significant problem because hyphae grew through the membrane and into the agar, and most transformants could be recovered after 7 days of incubation.
Hygromycin resistance conferred by hptII is commonly used as a selectable marker in Ascomycetes. However, spontaneous mutation to hygromycin resistance has been reported in M. fructicola (Dai et al. 2003), and also was found in the present study. Thus we used a cassette containing the ORF of nptII and the NOS terminator and promoter. NptII conferring G-418 resistance is a stable selectable marker and has been used for CaCl2/PEG-mediated/protoplast transformation of M. fructicola in separate experiments. In addition, a transformant carrying a single copy of integrated T-DNA carrying MFCUT1-GUS grew as well as the wild-type strain in SMM in the presence of 75 μg/ml G418 (M-H. Lee and R.M. Bostock, unpublished data).
We found that gene replacement through homologous recombination in M. fructicola occurred when 4 kb homologous regions of the Mfcut1 gene were used. Approximately 50% of the analyzed transformants harbored a disrupted copy of Mfcut1 in some nuclei. A similar frequency of homologous recombination and integration in M. fructicola also was observed using CaCl2/PEG-mediated/protoplast transformation with the same vector (M-H. Lee and R.M. Bostock, unpublished data).
Fungal transformation mediated by Agrobacterium can result in a single and/or multiple integrations of T-DNA (de Groot et al. 1998; Covert et al. 2001; Malonek and Meinhardt 2001; Leclerque et al. 2004; Mikosch et al. 2001; Hanif et al. 2002; Sullivan et al. 2002; Ahn et al. 2004; Michielse et al. 2005a, b). In a multinucleate fungus, a transformant may harbor mixed nuclei with various integration patterns. In addition, it is very likely that only few nuclei are competent to take up DNA. In N. crassa, DNA uptake and integration can only occur in some nuclei (Dev and Maheshwari 2002). The numbers of nuclei in an organism vary when grown on different media and conditions (Phillips et al. 1989; Davis 2000). In strain of M. fructicola used in our study, three nuclei per hyphal cell were usually detected when grown in SMM, while 8–11 nuclei per hyphal cell were detected when grown in V8 and YPD media. However, four rounds of single hyphal tip purification on SMM failed to identify homokaryotic Mfcut1 mutants of M. fructicola. Several attempts of single spore isolation on the SMM medium also were unsuccessful for obtaining a homokaryotic disruptant as assessed by PCR and Southern blot hybridization. Taken together, these results suggest that there is a nuclear preference of DNA transformation in M. fructicola MUK1.
The occurrence of T-DNA direct repeats in the genomes of fungi transformed by Agrobacterium has been reported (Mikosch et al. 2001; Rolland et al. 2003; Michielse et al. 2004a, b). However, no information on the frequencies of T-DNA direct and indirect repeat integration on fungi is available. In this study, T-DNA integrated as a series of direct repeats in the majority of transformants analyzed, with only a few cases of indirect repeat integrations detected (Figs. 6, 7, 8). In Arabidopsis, tomato and petunia, T-DNA integrates as indirect repeats more often than direct repeats (Jorgensen et al. 1987; Cluster et al. 1996; De Neve et al. 1997). However, T-DNA direct repeat integration occurs with higher frequency than indirect repeat integration in tobacco (Krizkova and Hrouda 1998). Host DNA repair mechanisms most likely control the T-DNA integration process (van Attikum et al. 2001; van Attikum and Hooykaas 2003). The high frequency of direct repeat integration in M. fructicola indicates that recombination mechanisms for DNA repair during T-DNA integration differ from those in plants.
Nucleotide deletions and substitutions occur frequently at the junctions of T-DNA repeats or insertion sites within host genomic DNA. In most plants, nucleotides in the left border (3′ end) more often are missing than in the right border (5′ end) of the T-DNA during integration (Tzfira et al. 2004). In Aspergillus awamori, transformation using Vir D2 mutants of Agrobacterium resulted in deletion of right border sequences more often than transformation of the wild-type strain (Michielse et al. 2004a, b). The right border is believed to be protected from exonucleases by VirD2 (Durrenberger et al. 1989; Mayerhofer et al. 1991; Bundock and Hooykaas 1996). However, two studies in tobacco show that nucleotides in the right borders are deleted more often than those in the left borders (Krizkova and Hrouda 1998; Salomon and Puchta 1998). In the present study, sequence analysis of the T-DNA junction fragment amplified from seven independent transformants indicated that the right borders (26 bp) at the junction of T-DNA direct repeats are missing in all of them, but only one in seven had a deletion in the 26 nucleotides of the left border. The 26 nucleotides of imperfect repeat border sequences in this study are present in pCAMBIA-1300 (Genbank accession No. AF-234296), which has been used as the backbone binary vector in Agrobacterium-mediated transformation of Fusarium oxysporum, Botrytis cinerea, and Beauveria bassiana (Mullins et al. 2001; Rolland et al. 2003; Leclerque et al. 2004). Among these studies, sequencing of the insertion sites within the genomic DNA showed that fewer nucleotides within the left border sequence were missing than within the right border sequence. This is consistent with our findings and suggests that among these fungi, including M. fructicola, VirD2 is removed before completion of the synthesis of the second strand of the T-DNA. Another possibility is that the T-DNA border regions from pCAMBIA-1300 are responsible for deletion of the nucleotides in the right border regions in all of these cases.
In conclusion, Agrobacterium-mediated transformation of M. fructicola provides an efficient method for generating transformants and also provides a system to study the DNA recombination system of this fungus. In N. crassa, the understanding of DNA recombination has provided information for generating a mutant with a “super-high” frequency of homologous gene replacement (Ninomiya et al. 2004). Because of the nature of multiple nuclei and the difficulty in obtaining homokaryotic disruptants, various approaches for gene functional analysis may need to be taken with M. fructicola, such as RNAi, or reporter gene analysis for studies of temporal and spatial expression. With respect to the latter, we have examined Mfcut1 expression during pathogenesis using an MFCUT1-GUS fusion (M-H. Lee and R.M. Bostock, unpublished results). Also, the use of a mononucleate stage such as ascospores or microconidia for transformation may increase the chances to obtain homokaryotic disruptants in this species. We have found that microconidia can express transgenes such as GFP and Mfcut1. However, procedures for induction of the sexual stage under laboratory conditions are not yet available and germination of microconidia has not been reported for this species. Establishing the germination requirements for M. fructicola microconidia, as has been done with N. crassa (Maheshwari 1999), could be very helpful for future research.
References
Ahn N, Kim S, Choi W, Im K-H, Lee Y-H (2004) Extracellular matrix protein gene, EMP1, is required for appressorium formation and pathogenicity of the rice blast fungus, Magnaporthe grisea. Mol Cells 17:166–173
Amey RC, Mills PR, Bailey A, Foster GD (2003) Investigating the role of a Verticillium fungicola beta-1,6-glucanase during infection of Agaricus bisporus using targeted gene disruption. Fungal Genet Biol 39:264–275
Asch DK, Kinsey JA (1990) Relationship of vector insert size to homologous integration during transformation of Neurospora crassa with the cloned am gdh gene. Mol Gen Genet 221:37–43
van Attikum H, Hooykaas PJJ (2003) Genetic requirements for the targeted integration of Agrobacterium T-DNA in Saccharomyces cerevisiae. Nucleic Acids Res 31:826–832
van Attikum H, Bundock P, Hooykaas PJJ (2001) Non-homologous end-joining proteins are required for Agrobacterium T-DNA integration. EMBO J 20:6550–6558
Bird D, Bradshaw R (1997) Gene targeting is locus dependent in the filamentous fungus Aspergillus nidulans. Mol Gen Genet 255:219–225
Bostock RM, Wilcox SM, Wang G, Adaskaveg JE (1999) Suppression of Monilinia fructicola cutinase production by peach fruit surface phenolic acids. Physiol Mol Plant Pathol 54:37–50
Bundock P, Hooykaas PJJ (1996) Integration of Agrobacterium tumefaciens T-DNA in the Saccharomyces cerevisiae genome by illegitimate recombination. Proc Natl Acad Sci USA 93:15272–15275
Bundock P, Den Dulk-Ras A, Beijersbergen A, Hooykaas PJJ (1995) Trans-kingdom T-DNA transfer from Agrobacterium tumefaciens to Saccharomyces cerevisiae. Embo J 14:3206–3214
Cluster PD, O’Dell M, Metzlaff M, Flavell RB (1996) Details of T-DNA structural organization from a transgenic Petunia population exhibiting co-suppression. Plant Mol Biol 32:1197–1203
Covert SF, Kapoor P, Lee M-H, Briley A, Nairn CJ (2001) Agrobacterium tumefaciens-mediated transformation of Fusarium circinatum. Mycol Res 105:259–264
Curtis KM (1928) The morphological aspect of resistance to brown rot in stone fruit. Ann Bot 42:39–68
Dai Q, Sun Z, Schnabel G (2003) Development of spontaneous hygromycin B resistance in Monilinia fructicola and its impact on growth rate, morphology, susceptibility to demethylation inhibitor fungicides, and sporulation. Phytopathology 93:1354–1359
Davis RH (2000) Neurospora: contributions of a model organism. Oxford University Press, New York, pp 333
de Groot MJA, Bundock P, Hooykaas PJJ, Beijersbergen AGM (1998) Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat Biotech 16:839–842
De Neve M, De Buck S, Jacobs A, Van Montagu M, Depicker A (1997) T-DNA integration patterns in co-transformed plant cells suggest that T-DNA repeats originate from co-integration of separate T-DNAs. Plant J 11:15–29
Dev K, Maheshwari R (2002) Transformation in heterokaryons of Neurospora crassa is nuclear rather than cellular phenomenon. Curr Micro 44:309–313
Dietz-Pfeilstetter A, Arndt N, Kay V, Bode J (2003) Molecular structure and regulatory potential of a T-DNA integration site in petunia. Transgenic Res 12:83–99
Durrenberger F, Crameri A, Hohn B, Koukolikova-Nicola Z (1989) Covalently bound Vir-D2 protein of Agrobacterium tumefaciens protects the T-DNA from exonucleolytic degradation. Proc Natl Acad Sci USA 86:9154–9158
Gelvin SB (2003) Agrobacterium-mediated plant transformation: The biology behind the “gene-jockeying” tool. Microbiol Mol Biol Rev 67:16–37
Gray M, Honigberg SM (2001) Effect of chromosomal locus, GC content and length of homology on PCR-mediated targeted gene replacement in Saccharomyces. Nuc Acids Res 29:5156–5162
Hanif M, Pardo AG, Gorfer M, Raudaskoski M (2002) T-DNA transfer and integration in the ectomycorrhizal fungus Suillus bovinus using hygromycin B as a selectable marker. Curr Genet 41:183–188
Hellens R, Mullineaux P, Klee H (2000) A guide to Agrobacterium binary Ti vectors. Trends Plant Sci 5:446–451
Jorgensen R, Snyder C, Jones JDG (1987) T DNA is organized predominantly in inverted repeat structures in plants transformed with Agrobacterium tumefaciens C58 derivatives. Mol Gen Genet 207:471–477
van Kan JAL, Van’t Klooster JW, Wagemakers CAM, Dees DCT, Van Der Vlugt-Bergmans CJB (1997) Cutinase A of Botrytis cinerea is expressed, but not essential, during penetration of Gerbera and tomato. Mol Plant Microbe Inter 10:30–38
Krizkova L, Hrouda M (1998) Direct repeats of T-DNA integrated in tobacco chromosome: characterization of junction regions. Plant J 16:673–680
Leclerque A et al (2004) Agrobacterium-mediated insertional mutagenesis (AIM) of the entomopathogenic fungus Beauveria bassiana. Curr Genet 45:111–119
Lee Y-W, Jin S, Sim W-S, Nester EW (1995) Genetic evidence for direct sensing of phenolic compounds by the VirA protein of Agrobacterium tumefaciens. Proc Natl Acad Sci USA 92:12245–12249
Longtine MS et al (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961
Maheshwari R (1999) Microconidia of Neurospora crassa. Fungal Genet Biol 26:1–18
Malonek S, Meinhardt F (2001) Agrobacterium tumefaciens-mediated genetic transformation of the phytopathogenic ascomycete Calonectria morganii. Curr Genet 40:152–155
Mayerhofer R, Koncz-Kalman Z, Nawrath C, Bakkeren G, Crameri A, Angelis K, Redei GP, Schell J, Hohn B, Koncz C (1991) T-DNA integration: a mode of illegitimate recombination in plants. EMBO J 10:697–704
Meyer V, Mueller D, Strowig T, Stahl U (2003) Comparison of different transformation methods for Aspergillus giganteus. Curr Genet 43:371–377
Michielse CB, Ram AFJ, Hooykaas PJJ, van den Hondel CAMJJ (2004a) Agrobacterium-mediated transformation of Aspergillus awamori in the absence of full-length VirD2, VirC2, or VirE2 leads to insertion of aberrant T-DNA structures. J Bact 186:2038–2045
Michielse CB, Ram AFJ, van den Hondel CAMJJ (2004b) The Aspergillus nidulans amdS gene as a marker for the identification of multicopy T-DNA integration events in Agrobacterium-mediated transformation of Aspergillus awamori. Curr Genet 45:399–403
Michielse CB, Arentshorst M, Ram AFJ, van den Hondel CAMJJ (2005a) Agrobacterium-mediated transformation leads to improved gene replacement efficiency in Aspergillus awamori. Fungal Genet Biol 42:9–19
Michielse CD, Hooykaas PJJ, van den Hondel CAMJJ, Ram AFJ (2005b) Agrobacterium-mediated transformation as a tool for functional genomics in fungi. Curr Genet 48:1–17
Mikosch TSP, Lavrijssen B, Sonnenberg ASM, van Griensven LJLD (2001) Transformation of the cultivated mushroom Agaricus bisporus (Lange) using T-DNA from Agrobacterium tumefaciens. Curr Genet 39:35–39
Mullins ED, Chen X, Romaine P, Raina R, Geiser DM, Kang S (2001) Agrobacterium-mediated transformation of Fusarium oxysporum: an efficient tool for insertional mutagenesis and gene transfer. Phytopathology 91:173–180
Ninomiya Y, Suzuki K, Ishii C, Inoue H (2004) Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. Proc Natl Acad Sci USA 101:12248–12253
Ogawa JM, English H (1991) Diseases of temperate zone tree fruit and nut crops. University of California, Division of Agriculture and Natural Resources, Oakland, Publication 3345
Phillips DJ, Margosan DA, Mackey BE (1989) Volume, nuclear number, and aggressiveness of conidia of Monilinia fructicola produced on media of varied glucose-concentrations at 15 C and 25 C. Phytopathology 79:401–403
Pratt RJ, Aramayo R (2002) Improving the efficiency of gene replacements in Neurospora crassa: a first step towards a large-scale functional genomics project. Fungal Genet Biol 37:56–71
Risseeuw E, Franke-Van Dijk MEI, Hooykaas PJJ (1996) Integration of an insertion-type transferred DNA vector from Agrobacterium tumefaciens into the Saccharomyces cerevisiae genome by Gap repair. Mol Cell Biol 16:5924–5932
Rolland S, Jobic C, Fevre M, Bruel C (2003) Agrobacterium mediated transformation of Botrytis cinerea, simple purification of monokaryotic transformants and rapid conidia-based identification of the transfer-DNA host genomic DNA flanking sequences. Curr Genet 44:164–171
Salomon S, Puchta H (1998) Capture of genomic and T-DNA sequences during double-strand break repair in somatic plant cells. EMBO J 17:6086–6095
Sullivan TD, Rooney PJ, Klein BS (2002) Agrobacterium tumefaciens integrates transfer DNA into single chromosomal sites of dimorphic fungi and yields homokaryotic progeny from multinucleate yeast. Euk Cell 1:895–905
Tinland B, Schoumacher F, Gloeckler V, Bravo-Angel AM, Hohn B (1995) The Agrobacterium tumefaciens virulence D2 protein is responsible for precise integration of T-DNA into the plant genome. EMBO J 14:3585–3595
Tzfira T, Li J, Lacroix B, Citovsky V (2004) Agrobacterium T-DNA integration: molecules and models. Trends Genet 20:375–383
Wang GY, Michailides TJ, Hammock BD, Lee Y-M, Bostock RM (2002) Molecular cloning, characterization, and expression of a redox-responsive cutinase from Monilinia fructicola (Wint.) Honey. Fungal Genet Biol 35:261–276
Acknowledgements
We thank Kuang-Ren Chung, Steve Daubert, Lynn Epstein, Tom Gordon, and John Labavitch for their critical review of the manuscript, and Steve Marek, Clarence Kado and Jeffrey Rollins for their helpful discussions. We also thank Dan Johnson, Sheilah Krishnan, and Betsy Harbert for their help with fungal genomic DNA isolation. Research supported in part by grants from the California Cling Peach Board and the US-Israel BARD program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Heitman
Note: Nucleotide sequence data reported are available in GenBank under the accession number–DQ173196.
Rights and permissions
About this article
Cite this article
Lee, MH., Bostock, R.M. Agrobacterium T-DNA-mediated integration and gene replacement in the brown rot pathogen Monilinia fructicola . Curr Genet 49, 309–322 (2006). https://doi.org/10.1007/s00294-006-0059-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-006-0059-0