
Abstract
Mutagenesis was used to study the function by the ALR1 (aluminium resistance) gene, which encodes the major Mg2+ uptake system in yeast. Truncation of Alr1 showed that the N-terminal 239 amino acids and the C-terminal 53 amino acids are not essential for magnesium uptake. Random PCR mutagenesis was undertaken of the C-terminal part of ALR1 that is homologous to the bacterial CorA magnesium transport family. The mutants with the most severe phenotype all had amino acid changes in a small region containing the putative transmembrane domains. Eighteen single amino acid mutants in this critical region were classified into three categories for magnesium uptake: no, low and moderate activity. Seventeen of the 18 mutants expressed a cross-reacting band of similar size and intensity as wild-type Alr1. Conservative mutations that reduced or inactivated uptake led us to identify Ser729, Ile746 and Met762 (part of the conserved GMN motif) as critical amino acid residues in Alr1. High expression of inactive mutants inhibited the capability of wild-type Alr1 to transport magnesium, consistent with Alr1 forming homo-oligomers. The results confirm the classification of ALR1 as a member of the CorA family of magnesium transport genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Magnesium is the most abundant divalent cation in eukaryotic and prokaryotic cells. Intracellular Mg2+ concentration is regulated and appears to be maintained by regulation of uptake, transport into organelles (ER, vacuole etc.) or Mg2+ efflux. Despite its importance, little is known about the mechanism of Mg2+ transport and the regulation of Mg2+ homeostasis in cells (Gardner 2003).
The CorA family of Mg2+ transport genes are found in prokaryotic and eukaryotic cells (Drummond et al. 2005). In Salmonella typhimurium and Escherichia coli, the CorA gene (Cobalt resistance) encodes the primary Mg2+ transport system (Hmiel et al. 1986; Grubbs et al. 1989). The CorA gene family is widely distributed throughout Bacteria and Archaebacteria (Kehres et al. 1998), and a subset of this family is found in fungi (MacDiarmid and Gardner 1998; Gardner 2003; Drummond et al. 2005). A related sub-family of genes, the MRS2 gene family (for mitochondrial RNA splicing) is involved in Mg2+ uptake into mitochondria of both yeast (Bui et al. 1999) and mammals (Zsurka et al. 2001). In plants, the MRS2 family has been amplified and consists of ten genes in Arabidopsis thaliana (AtMRS2 genes), three of which complement Mg2+ uptake in bacteria and/or yeast (Schock et al. 2000; Li et al. 2001; Drummond et al. 2005).
The CorA protein has three transmembrane (TM) domains near the C-terminus, based on protein fusions in E. coli (Smith et al. 1993). A large, highly charged, N-terminal region is found in the periplasm. However, the related yeast mitochondrial transport protein, Mrs2 (Bui et al. 1999), and a variant CorA protein involved in Zn2+ efflux in enteric bacteria, ZntB (Worlock and Smith 2002; Caldwell and Smith 2003) have only two TM domains. CorA probably functions as a homo-tetramer (Warren et al. 2004), while Mrs2 forms a homo-pentamer (Kolisek et al. 2003). All CorA family members have a highly conserved GMN motif located near the external face of the penultimate TM domain (TM1 in Mrs2, TM2 in CorA). This sequence is essential for function (Szegedy and Maguire 1999) and forms the signature motif of this protein family.
Two members of the CorA family in yeast (Saccharomyces cerevisiae) are called ALR1 and ALR2, because overexpression of the genes confers an aluminium resistant phenotype. ALR1 encodes the major Mg2+ uptake protein in laboratory strains of S. cerevisiae (MacDiarmid and Gardner 1998; Graschopf et al. 2001); ALR2 appears to be functionally equivalent to ALR1 but is not transcribed in strain S288C (MacDiarmid and Gardner 1998). ALR1 mRNA is induced at low external Mg2+ concentrations and the Alr1 protein is subject to internalisation and degradation in high external Mg2+ (Graschopf et al. 2001). A patch clamp study in yeast indicates Alr1 may mediate both Mg2+ influx and efflux and likely acts as a channel (Liu et al. 2002). A number of other divalent cations are substrates for the Mg2+ uptake system, including Co2+, Ni2+, Mn2+ and Zn2+ (Fuhrmann and Rothstein 1968; MacDiarmid and Gardner 1998). In yeast Mg2+ uptake via Alr1 is inhibited by aluminium (MacDiarmid and Gardner 1996, 1998).
The topology of the Alr1 protein is not known. Computer algorithms predict two TM domains near the C terminus. However, the same algorithms predict only two TM domains in CorA, whereas the experimental data suggest that there is a third domain immediately upstream (Smith et al. 1993). Alr1 may therefore have three TM domains with a large N-terminal domain located externally (like CorA) or it may have only two TM domains like Mrs2 and ZntB, with the N-terminal domain located internally. Alr1 is much larger than CorA (95 kDa compared to 37 kDa). Although overall amino acid similarity between members of the family is low, a CorA protein domain has been defined by the PFAM and conserved domain databases (see Drummond et al. 2005). The CorA domain includes an N-terminal region that is rich in non-polar amino acids and is also highly charged and a C-terminal region with two or three TM domains, one with the GMN motif. The CorA domain in Alr1 is located towards the C terminus of the protein (amino acid residues 451–803, as defined by PFAM family PF01544), with large N-terminal and short C-terminal extensions; these N- and C-terminal extensions are shared in most of the other fungal homologues.
Mutational studies on TM2 (Szegedy and Maguire 1999) and TM3 (Smith et al. 1998) of the CorA bacterial gene revealed that both are essential for Mg2+ transport. In particular, changes to the highly conserved GMN motif in TM2 of CorA abolished Mg2+ uptake, and amino acids with hydroxyl side chains (T, Y, S) in both TM domains were also found to be important for activity. There is only one negatively charged amino acid (in TM1), which was not required for transport (Smith et al. 1998).
In this study, we present a mutational analysis of the yeast ALR1 gene aimed at defining the regions of the protein that are critical for Mg2+ uptake. Deletion analysis was used to see whether the N-terminus and C-terminus of Alr1, which are located outside of the CorA domain, are essential for Mg2+ uptake. Random mutagenesis was used to identify critically important regions and residues within the CorA domain. The effect of overexpressing the inactive mutations on the function of wild-type Alr1 was also investigated, to assess whether the proteins may assemble into homo-oligomers.
Materials and methods
Yeast strains and media
Yeast strains CM52 (MATa his3-△200 ura3-52 leu2-△1 lys2△202 trp1△63) and CM66 (MATa alr1::HIS3 alr2::TRP1 his3-△200 ura3-52 leu2-△1 lys2△202 trp1△63) were used in this work (Li et al. 2001). The hac1, ire1 or his1 deletion mutants in the background of the diploid strain BY4743 (MATa/α his3 leu2 met15 lys2 ura3) were purchased from Research Genetics (Saccharomyces Genome Deletion Project). Yeast was cultivated in yeast extract peptone dextrose (YPD) (Sherman 1991), YPD + high Mg2+ YPDM (YPD + 250 mM MgCl2) and standard synthetic medium (SC-ura, synthetic complete medium without uracil, Sherman 1991) with the appropriate Mg2+ content added. Low pH low Magnesium (LPM) media (pH 3.5, Mg2+ 200 μM) was developed for analysis of aluminium tolerance (MacDiarmid and Gardner 1996). Here we used LPM-ura (LPM media lacking uracil) both for growth on low Mg2+ and for metal tolerance tests with added aluminium (70 μM), cobalt (500 μM) or nickel (500 μM). All strains were grown at 30°C. Standard SC-ura contained 2 % glucose; alternative carbon sources were sometimes added when the GAL1 promoter was used (see text).
Cloning and plasmid manipulation
The ALR1 coding region, together with its native promoter, was isolated as a DraI fragment from a larger clone of the genomic region (C. MacDiarmid, unpublished data). This fragment was ligated into the low-copy pFL38 vector (ARS/CEN, URA3) (Bonneaud et al. 1991) that had been digested with SmaI and SphI (Invitrogen), made blunt-ended with Klenow (Invitrogen) and dephosphorylated with shrimp alkaline phosphatase (Roche). This step generated pFL38–ALR1. HA-tagged proteins in this vector were derived using two plasmid templates from Longtine et al. (1998): pFA6a-3HA–KanMX6 (C-terminal fusions driven by the native ALR1 promoter) and pFA6a–KanMX6–pGAL1-3HA (N-terminal fusions driven by the GAL1 promoter). The two HA-Kan cassettes were amplified by polymerase chain reaction (PCR) using the oligonucleotides listed in Table 1 and recombined in vivo by co-transforming them with pFL38–ALR1 into strain CM66; colonies were selected for functional Mg2+ transport and geneticin resistance (by growing them on SC-ura with 4 mM Mg2+ and 200 mg/l geneticin). Sequencing confirmed the successful recombination of the 3xHA epitope into the C terminus or N terminus of Alr1; the resulting plasmids were designated as pFL38–ALR1-HA and pFL38–pGAL1-HA–ALR1, respectively.
Plasmid pFA6a–KanMX6–pGAL1-3HA was also used as a template for generating N-terminal truncated mutants of Alr1 (designated as Del 1 through Del 4), while pFA6a-3HA–KanMX6 was used as a template for generating the C-terminal truncation (Del 5). The HA-Kan and Kan–pGAL1-HA cassettes were amplified by PCR using the listed oligonucleotides (Table 1). The PCR products were recombined in vivo into pFL38–ALR1 in CM52 (with selection for geneticin resistance but not for magnesium uptake) and the products confirmed by sequencing as above.
PCR mutagenesis
Random PCR mutagenesis of pFLN2–ALR1 (Li et al. 2001) was used to identify crucial amino acids in the 3′ half of the ALR1 gene (amino acids 465–859). Standard PCR was performed as follows: 200 pg of pFLN2–ALR1 template, 1x PCR reaction buffer (20 mM Tris–HCl [pH 8.4], 50 mM KCl), 200 μM each dNTP, 0.2 μM of each primer (1/5 and LACR, Table 1), TaqI polymerase (2.5 U), 1.5 mM MgCl2 (50 μl total volume); the reaction was incubated at 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 30 s, followed by 72°C for 7 min. The PCR products were recombined in vivo into pFLN2–ALR1, by co-transforming (Muhlrad et al. 1992; Gietz et al. 1995) the PCR product with pFLN2–ALR1 plasmid that had been digested with PstI and NotI. Ura+ transformants in strain CM66 (alr1alr2) were grown on medium non-selective for magnesium (SC-ura supplemented with 250 mM Mg2+) and individual colonies were screened for reduced growth on normal Mg2+ (SC-ura 4 mM) or low Mg2+ (LPM-ura 200 μM). Some plasmids were selected that showed wild-type growth on magnesium but had both reduced tolerance to aluminium, and increased tolerance to cobalt and nickel, a phenotype expected for low Alr1 activity (MacDiarmid and Gardner 1998). Plasmid DNAs were prepared from candidate mutants and transformed into E. coli by electroporation. The plasmids were retransformed into CM66, retested on the same media to confirm their phenotype and sequenced.
The putative TM-domain region of some of these mutants was transferred into the new vector, pFL38–ALR1-HA as follows. The mutant plasmids were digested with ClaI and NotI and the targeted band was excised and purified. These purified fragments were recombined into the new vector pFL38–ALR1-HA cut with XbaI and MscI in CM66, and cycled through E. coli back into CM66 as above. Successful recombination of the single amino acid changes was confirmed with sequencing.
Manganese-mediated PCR mutagenesis was used to mutate the putative TM-domain region (from amino acid 657 to 859) of pFL38–ALR1-HA. PCR was performed as above, except that the template was pFL38–ALR1-HA, the primers were 1/10 and HM–Alr1 (Table 1), and the reaction was undertaken in the presence of either 25 μM or 50 μM Mn2+ (Svetlov and Cooper 1998). The PCR product was recombined in vivo by co-transformation into CM66 with pFL38–ALR1-HA digested with XbaI and MscI. Mutant screening on low Mg2+, transformation into E. coli, retesting of the phenotype in CM66 and DNA sequencing were as above.
Growth measurements
CM66 yeast strains with wild-type and mutant plasmids were grown to saturation on liquid SC-ura media with 250 mM Mg2+. The cells were harvested by centrifugation (800 g), washed with distilled water three times and resuspended in fresh SC-ura media (no added Mg2+). Aliquots of the cells were diluted (40–50-fold) into SC-ura media containing different Mg2+ concentrations to give a final OD600 = 0.05. The OD600 of the cultures was followed over 72 h.
Measurement of Mg2+ content
CM66 yeast strains with wild-type and mutant plasmids were grown, harvested and washed as above. The cells were diluted to OD600=0.5 in fresh SC-ura media (no added Mg2+) and starved by incubating them for 24 h. The cells were collected, resuspended and incubated with SC-ura with 1 mM Mg2+ for 2 h; 10 ml aliquots were sampled over time. The cells were washed three times, with distilled water, 1 mM EDTA and distilled water respectively. The harvested cells were resuspended in 2 ml of distilled water. A 0.25-ml sample was taken for measurement of OD600 and the remaining sample (1.75 ml) was mixed with 1.75 ml of concentrated nitric acid. After incubation at 100°C for 1 h, samples were mixed with 2x LaCl3 buffer (250 mM HCl, 47 mM LaCl3). The Mg2+ contents of samples were measured with either a Varian 1275 (Palo Alto, CA, USA) or an Avanta (GBC, AU) atomic absorption spectrophotometer, using an air-acetylene flame at a wavelength of 285.2 nm.
Western blot analysis
CM66 yeast strains with wild-type and mutant plasmids driven by the native ALR1 promoter were grown to saturation in liquid SC-ura media containing 250 mM Mg2+, centrifuged and washed three times in water as above. They were then starved for 24-h period in SC-ura medium without Mg2+ prior to protein analysis. For plasmids driven by the GAL1 promoter, cells were grown for 24 h on SC-ura media (with 0.2% glucose, 1.8% galactose and 0.9% raffinose) supplemented with either 10 μM Mg2+ (pFL38–pGAL1-HA–ALR1, Del 1 and 2) or 4 mM Mg2+ (Del 3 or Del 4). The cells were collected by centrifugation, washed with distilled water three times and total proteins were prepared using the post-alkaline method of Kushnirov (2000). Proteins were resolved by 8% SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membrane at 35 V overnight in a cold room. Western blots were performed using mouse anti-HA monoclonal antibody from Roche (1:2,000) and Goat anti-mouse secondary antibody (1:3,000, Biorad). Antibody detection was carried out according to the ECL protocol from Amersham Bioscience. The membranes were exposed to Kodak X-OMAT K film and developed with AGFA Curix 60 developer. The membranes were stripped (following instructions of the supplier, Pierce) and incubated with anti-H+ATPase (Pma1) antibody (1:20,000) (Serrano et al. 1993) to estimate lane loadings. All Western blots were repeated at least once.
Results
The N- and C-termini of Alr1 are not essential for Mg2+ uptake
The yeast Alr1 protein (859 amino acids) is much larger than bacterial CorA proteins (for example, the S. typhimurium protein has 316 amino acids). The CorA domain is located towards the C-terminus, with Alr1 having a large N-terminal extension (around 400 amino acids) and a short C-terminal extension (around 60 amino acids) relative to CorA. No clear function of these termini is indicated by database searches for homologous proteins (data not shown). Deletion mutagenesis was used to examine the functional role of these regions in Mg2+ transport.
Four N-terminal truncated mutants were constructed, each fused to HA and driven from the GAL1 promoter. CM66 cells expressing proteins lacking up to 239 amino acids from the N-terminus (Del 1 and Del 2, Fig. 1a) were able to grow on plates containing 4 mM Mg2+, but two larger N-terminal deletions (Del 3 and Del 4, Fig. 1a) abolished growth on 4 mM Mg2+ (cells grew similarly to wild-type on 250 mM Mg2+, data not shown). The result was independent of whether the deletion mutants were grown on glucose or galactose (Fig. 1a). Our previous results with the GAL1 promoter suggest that it directs a reasonable level of transcript expression in glucose (Ezaki et al. 1999).

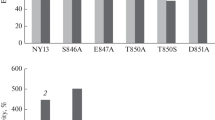
The N- and C-termini of Alr1 are not crucial for Mg2+ uptake by yeast. a The deletion mutants (illustrated on the left) were tested for their ability to grow on 4 mM Mg2+ (right side of figure) with either 2% glucose or 2% galactose. CM66 cells expressed either the wild-type gene (pFL38–pGAL1-HA–ALR1), empty vector (pFL38), N-terminal deletion mutants (Del1 through Del4 in pFL38–pGAL1-HA–△ALR1; numbers refer to the first amino acid residue present in the deleted Alr1 protein) or C-terminal deletion mutants (Del5△, frame shift occurred after amino acid 806, followed by the amino acids PDPRVN; Del5, 3x HA fused to amino acid 806). The wild-type and Del1--Del4 were all driven by the GAL1 promoter whereas expression of Del5△ and Del5 was from the endogenous ALR1 promoter. Black boxes represent the two TM domains predicted by computer algorithms and grey box represents putative TM1, based on homology to CorA. b Magnesium uptake into starved CM66 yeast cells expressing various deleted ALR1 genes was measured after 0 and 2 hours using AAS in SC-ura media containing 1 mM Mg2+. The column graph shows the relative uptake (%) measured as the increase in Mg2+ contents between the starting point (0 h) and the end point (2 h). Points are the average (±SE) derived from three independent experiments. c All the deletion mutants express the HA epitope as part of a fusion product of the expected size. The five left-most lanes are from cells expressing HA–ALR1 fusions driven by the GAL1 promoter, and were cultured on SC-ura (with 0.2% glucose, 1.8% galactose and 0.9% raffinose) containing either 10 μM Mg2+ (pFL38--pGAL1-HA–ALR1, Del 1 and 2) or 4 mM Mg2+ (Del 3 or Del 4). The last two lanes contain ALR1-HA driven by the native ALR1 promoter; these cells were cultured on SC-ura media with 250 mM Mg2+ and then starved for Mg2+ for 24 h. The mobilities of co-electrophoresed MW markers are indicated. The expected size of Alr1 is 100 kDa, and the five deletion mutants are expected to be 81, 73, 64, 56 and 94 kDa respectively
Direct measurement of Mg2+ uptake was carried out using the four deletion mutants and compared to wild-type ALR1. The cells were starved for Mg2+ for 24 h prior to measuring uptake, to equalise their internal Mg2+ stores (Graschopf et al. 2001). Mg2+ uptake by the mutant proteins was followed over 2 h in minimal defined media with 1 mM Mg2+. Figure 1b shows that the cells containing wild-type plasmid increased their Mg2+ content by 60% over 2 h. The first two deletion mutants (Del1, Del2), lacking up to 239 N-terminal amino acids, were able to increase intracellular Mg2+ to the same extent as wild-type. In contrast, the other two larger N-terminal deletions (Del3, Del4) were unable to take up Mg2+, as were cells with an empty vector (see Fig. 4). These findings present direct evidence for the dispensability of the N-terminal 239 residues in Alr1 for Mg2+ uptake.
Figure 1c shows that proteins were expressed from each of the deletions of the expected size compared to the wild-type (100 kDa). In some cases an additional band was seen around 20 kDa larger than that of the expected size, whose origin is not clear; we note that a larger-than-expected band was also evident in the results of Graschopf et al. (2001).
Alr1 has 62 residues after the predicted end of the last transmembrane (TM) domain compared to 6 residues in CorA. A C-terminal deletion mutant was constructed that included the first 806 amino acid residues from Alr1 fused with a C-terminal HA epitope. Two candidate mutants were sequenced. The first C-terminal deletion (Del5△) contained an unexpected frame shift in the region of the homologous recombination, which resulted in a protein containing 806 residues from Alr1, with six additional amino acid residues (PDPRVN, encoded by the out-of-frame HA epitope) followed by a stop codon (this mutant therefore did not express the HA epitope). The second mutant (Del5) contained the expected construct, with 806 residues of Alr1 fused to the 3xHA epitope.
The first Del5△ mutant grew similarly to wild-type on plates containing 4 mM Mg2+ (Fig 1A) as well as on 200 μM (data not shown). Direct measurement of Mg2+ uptake showed that it exhibited wild-type levels of Mg2+ uptake (Fig. 1b). These results demonstrate that lack of the C-terminus did not cause any reduction of Mg2+ uptake. However, the second Del5 mutant showed a severe growth defect on normal Mg2+ plates (Fig. 1a) and provided no capability for Mg2+ uptake into CM66 cells over 2 h (Fig. 1b). The reduction in phenotype was not due to lack of expression, because Western analysis showed the presence of a HA-reactive band of about the expected size and intensity (Fig. 1c).
The basis for the difference in phenotype between these two deletion mutants is not clear. The addition of the HA epitope to wild-type Alr1 causes a reduction in Mg2+ uptake (see below), but not to the extent evident between these two mutants. The high activity of the first “frame shifted” mutant clearly showed that the C-terminus of Alr1 is not essential for Mg2+ uptake activity. It may be that the relatively close proximity of the HA epitope to the TM domains inhibited uptake in some way.
We conclude that expression of a functional Mg2+ transporter in yeast requires a region that is somewhat larger than the CorA domain. Part of the N-terminal region (amino acids 1–239) and the C terminal 53 amino acids appear not to be absolutely essential to obtain good growth on 4 mM Mg2+. These regions of the Alr1 protein may play additional roles in yeast.
Mutants with reduced Mg2+ transport cluster in the TM domain region of Alr1
The region of the ALR1 gene initially targeted for PCR mutagenesis was the 3′ half of the gene (1.2 kb, encompassing amino acids 465–859), which contains the majority of the CorA domain. A series of 20 mutants were isolated and analysed in a high copy expression vector, pFLN2–ALR1. These mutants each had one to three amino acid changes and showed phenotypes on plates that suggested reduced Mg2+ uptake (data not shown). The eight mutants with the most severe phenotypes showed growth that was indistinguishable from empty vector. All of these severe mutants had an amino acid change within a small region of the protein containing the putative TM domains (in four cases these were single amino acid changes). The reduced growth did not seem to result from the mis-trafficking of the mutants because GFP fusions constructed with three of the single amino acid mutants localised to the plasma membrane (data not shown). This initial mutant screen therefore identified this localised region of the protein (which included amino acid residues 716 to 789) as being critical for magnesium uptake.
During this initial round of mutagenesis, we noted inconsistencies in the data that appeared to result from variation between colonies in the level of expression directed from the pFLN2 vector. We therefore constructed a new low-copy (pCEN) vector in which the ALR1 gene was expressed from its own promoter. This plasmid, pFL38–ALR1-HA, incorporated three copies of the haemaglutenin (HA) epitope at the C-terminus of Alr1. The addition of the HA epitope at the C-terminus of Alr1 somewhat decreased the capacity of the protein to confer growth at low Mg2+ levels. Measurements in liquid medium showed a reduction in growth at 10 μM Mg2+ and the relative colony size was reduced on LPM (200 μM Mg2+) and on media containing aluminium, compared to cells with the wild-type gene (data not shown).
Ten of the original mutations obtained in the initial screen above, including seven of the eight most severe mutants, were transferred individually to the pFL38–ALR1-HA vector. With one exception (Fig. 2), they showed similar phenotypes in the new vector. A second round of mutagenesis using manganese was also undertaken in this vector, focused on the smaller C-terminal region that had proved to be critical for Alr1 function in the first mutagenesis. Eight new missense mutants were selected, each with a single amino acid change.

The mutants show reduced growth on plates with low Mg2+. CM66 cells expressing wild-type (pFL38–ALR1-HA), empty vector (pFL38) or individual mutants were grown in SC-ura supplemented with 250 mM Mg2+, serially diluted 20-fold twice and plated on a SC-ura supplemented with 250 mM Mg2+, or b LPM-ura medium containing 200 μM Mg2+. The 18 mutants were classified as “no growth”, “intermediate growth” or “wild-type growth” (mutants in each class are listed in Fig. 3). The mutants are arranged in order of their location; putative TM locations are given in Fig. 7. Asterisks indicate mutants generated by mutagenesis of pFL38–ALR1-HA. One (S729P) of the mutants transferred from the 2 μm plasmid, showed a relatively more severe phenotype in the pFL38 vector, presumably because of lower expression. SC plates with 4 mM Mg2+ were also used and gave very similar results to the LPM plates. The LPM plate results are shown in b because the lower magnesium concentration (200 μM) illustrates the differences in the ïntermediate¨ and ¨wild-type¨ class of mutants more clearly
The growth of these eighteen mutants on plates with low Mg2+ is shown in Fig. 2. The mutants fell into three categories. The most severe class (termed “no growth”) showed growth on low Mg2+ that was indistinguishable from empty vector. The ten mutants in this category included M762L, in which the methionine residue of the conserved GMN motif was mutated to leucine. The other classes of mutants showed “intermediate growth” (four mutants) or growth that was “wild-type” or slightly reduced (four mutants). This latter category of mutants also showed reduced growth on aluminium and decreased sensitivity to cobalt (data not shown), phenotypes indicative of a reduced capacity for magnesium uptake (MacDiarmid and Gardner 1998). Two of the new mutants (G694C and L701F) apparently occurred as a result of changes in the region of crossing over in the recombination step.
In addition to the eight missense mutants, three nonsense mutations were obtained in the second mutagenesis, all of which showed no growth on low Mg2+ (data not shown). These were located after amino acids 722, 734 and 769. In unrelated selection experiments, we obtained downstream nonsense mutants (after amino acids 814, 816, 823, 845) that showed wild-type growth on LPM plates (200 μM Mg2+, data not shown). The region between amino acids 769 (the last inactive nonsense mutant) and 814 (the first active nonsense mutant) must therefore contain the C-terminal end of the essential region. These results are entirely consistent with the data presented above, in which the C-terminal boundary of the essential region was further localised by the identification of an essential residue at position 789 (Fig. 2 and 3), and the finding that a mutant deletion of residues after 807 is active (Fig. 1).

The mutants show reduced growth rates in liquid medium with low Mg2+. CM66 cells expressing wild-type (pFL38–ALR1-HA), empty vector (pFL38) or individual mutants were grown to saturation on liquid SC-ura media containing 250 mM Mg2+. The cells were harvested, washed with distilled water three times and diluted 40--50-fold into SC-ura media containing different levels of Mg2+ (20, 200 μM, 1, 4, 10, 250 mM) to give a final OD600=0.05. Growth rates over 72 h are shown for: a wild-type (pFL38–ALR1-HA) and empty vector (pFL38); b “no growth” mutants showing a pattern similar to empty vector; c “intermediate growth” mutants showing growth rates between wild-type and empty vector; d “wild-type growth” mutants showing reduced growth only on very low (20 μM) Mg2+. Points are the average (±SE) derived from three independent experiments
The mutations cause reduced magnesium uptake by Alr1
Growth rates of the mutants were measured in liquid media containing different levels of Mg2+. The empty vector (pFL38) is unable to grow at normal rates even with 10 mM Mg2+ in the medium (Fig 3a). Ten mutants classified as “no growth” on plates all showed growth patterns in liquid that were indistinguishable from cells containing the empty vector (Fig. 3b). We noted that cells with the N754L mutant showed a growth defect compared with empty vector even at high Mg2+; the reason for this effect on growth is unclear. The four mutants categorised as “intermediate growth” on plates also showed intermediate growth rates in liquid for most Mg2+ levels, when compared to cells expressing wild-type ALR1 gene (Fig. 3c). The last four mutants, originally classified as “wild-type” growth on plates, showed wild-type growth at higher Mg2+ concentrations (200 μM to 10 mM) but their growth was reduced at the lowest Mg2+ concentration tested (20 μM; see Fig. 3d) compared with wild-type pFL38–ALR1-HA (Fig 3a). The mutant classification of the low copy number mutants based on liquid media (Fig. 3) was therefore completely consistent with the solid media results (Fig. 2).
Direct measurement of Mg2+ uptake was undertaken using one mutant from each class (M762L, S729T, A774S) and compared to wild-type ALR1 or empty vector. Figure 4 shows that the cells containing wild-type plasmid increased their Mg2+ content by 45% over 2 h, while cells with empty vector showed no Mg2+ uptake. We note that this is a lower rate of Mg2+ uptake than previous results (Graschopf et al. 2001). The difference is in part due to the presence of the HA epitope, because pFL38--ALR1 without HA increased Mg2+ content by 75% in 200 μM Mg2+ (data not shown). We also note that HA fusion into the N-terminus provided no significant effect on Mg2+ uptake compare to wild-type (Fig. 1b)

The mutants show decreased uptake of Mg2+ into the cell Magnesium uptake into starved CM66 yeast cells expressing various ALR1 genes was measured over 2 h (0, 20 min, 1 h and 2 h) using AAS in SC-ura media containing 1 mM Mg2+. pFL38–ALR1-HA (wild-type), solid squares; A774S (“wild-type growth” mutant), open squares; S729T (“intermediate growth” mutant), solid triangles; M762L (“no growth” mutant), open triangles; empty vector (pFL38), solid circles
The three mutants tested showed varying levels of Mg2+ uptake that correlated with their growth phenotype. Thus the “no growth” mutant, M762L, showed essentially no Mg2+ uptake, suggesting that the alteration of this amino acid resulted in abolished Mg2+ uptake. Cells expressing the “intermediate growth” mutant, S729T, and the “wild-type growth” mutant, A774S, exhibited intermediate rates of uptake of 1 mM Mg2+.
Based on this correlation between growth rates in limiting magnesium and rates of magnesium uptake measurement, we will refer to the three phenotypic classes of mutants as “no activity”, “low activity” and “moderate activity” respectively.
Alr1 protein is expressed in the mutants
To determine if the mutations had any effect on Alr1 protein stability or steady state expression, Western analysis was used to monitor protein expression of the mutants. Graschopf et al. (2001) previously showed that at high external Mg2+, wild-type Alr1 protein is internalised and degraded. Initial western blots confirmed that high external Mg2+ (250 mM) reduced the amount of cross-reacting HA epitope from the pFL38–ALR1-HA plasmid (data not shown). However, to obtain sufficient cells of the inactive mutants for analysis, all cultures had to be grown up at 250 mM Mg2+. To circumvent this problem, we subjected all cultures to a 24-h period of Mg2+ starvation prior to protein extraction. The results, presented in Fig. 5, show that the wild-type and all the mutant plasmids expressed a band of the expected size (ca. 100 kDa). A larger-than-expected additional band (at 120 kDa) was again evident in some lanes. There was some variation between samples in the amount of cross-reacting 100-kDa protein seen; however, all the mutants except G790D showed a cross-reacting band whose intensity was similar to the wt protein or higher (Fig. 5). We therefore conclude that, for 17 of the 18 mutants, the loss of Mg2+ uptake activity was not due to a reduction of protein expression. The moderate activity mutant, G790D, showed the presence of a cross-reacting band, but it was present at a lower intensity than wild-type Alr1 (in Fig. 5, as well as in two other experiments in which it was grown in 4 mM Mg2+ without a starvation period). In this case the reduced activity of the mutant may be a result of a lower level of protein expression or mis-processing of the protein.

All the mutant proteins are expressed. CM66 cells expressing wild-type (pFL38–ALR1-HA), empty vector (pFL38) or individual mutants were grown up in 250 mM Mg2+ and then starved for Mg2+ for 24 h. Total proteins were prepared and Western blots were carried out to analyse HA or Pma1 proteins, as indicated. a “no activity” mutants b “low activity” and “moderate activity” mutants. The predominant cross-reacting band against HA migrated similarly to the 100-kDa marker, while that against Pma1 migrated above the 190-kDa marker
High-level expression of inactive mutants causes a dominant negative phenotype
Four single mutants (S729P, M762L, S771P, L783P) were introduced into CM52, which contains a wild-type chromosomal copy of ALR1. The mutants were either expressed at high level from the pFLN2 vector, or at a lower level from the pCEN vector pFL38 (with HA fusions). High-level expression of all four mutants inhibited the capability of the wild-type gene to support growth, even on the comparatively high level of 4 mM Mg2+. The results for two mutants are shown in Fig. 6a. The growth inhibition was overcome by adding 250 mM Mg2+ (compare growth of the overexpressed mutant on 250 mM Mg2+ with that of the CM66 containing vector only). This result suggests that overexpression of the mutant protein induces a specific negative effect on the expression or activity of the wild-type Alr1 protein, rather than a non-specific effect on cell growth. Low-level expression of the mutants from the pCEN vector did not affect the activity of the wild-type gene at 4 mM Mg2+.

Inactive mutants of Alr1 show a dominant negative phenotype a Two inactive mutants (M762L, S771P) and wild-type ALR1 gene, cloned in either a high copy vector (pFLN2) or a low copy vector (pFL38), as well as empty vector (pFLN2) were transformed into CM52 (ALR1 ALR2). Cells were grown on SC-ura media supplemented with 250 mM Mg2+ and five-fold serial dilutions plated on media containing high (250 mM) or low (4 mM) Mg2+. For comparison, CM66 with pFLN2 is shown (bottom). b One inactive mutant (M762L) and the wild-type ALR1 gene expressed from pFLN2, as well as empty vector (pFLN2), were transformed into a diploid strain of yeast (BY4743, wild-type for ALR1 ALR2) with deletions in either the HAC1, IRE1 or HIS1 genes (the latter was used as a wild-type strain). Cells were grown, diluted and plated as above
The results are consistent with the hypothesis that the active form of Alr1 is an oligomer, and that binding of the mutant to the wild-type protein results in an inactive complex. Alternatively, proteolytic degradation of the oligomeric complex may be involved, since the dominant negative phenotype was only observed following overexpression. Casagrande et al. (2000) showed that overexpression of a membrane protein can induce its degradation via the unfolded protein response (UPR) in the endoplasmic reticulum (ER). To exclude this explanation, we utilised hac1 and ire1 mutant lines, which are defective in the UPR (Patil and Walter 2001; Lee et al. 2003). The dominant negative growth effect of M762L was observed in both the hac1 and ire1 deletion mutant lines (Fig. 6b) and the effect was of similar magnitude in the wild-type strain as in either mutant. In all strains the growth effect was completely reversed at 250 mM Mg2+ (compare to CM66, bottom row of Fig. 6a). We conclude that the dominant negative effect of the Alr1 mutants occurs independently of the UPR. Moreover, the lack of any indication of synthetic lethality between ire/hac mutants and mutant overexpression indicates that this overexpression of Alr1 probably does not involve ER stress (Casagrande et al. 2000).
Discussion
Part of the large N terminus and the small C-terminus are not essential for Mg2+ uptake
The Alr1 protein is larger than the CorA bacterial protein (95 kDa compared to 37 kDa), with a significant N-terminal extension. Two N-terminal truncations showed that amino acids 1–239 of Alr1 are dispensable for Mg2+ uptake, but two larger deletions (encompassing amino acids 240–396) showed significant reduction of growth and Mg2+ uptake at normal Mg2+. We conclude that effective magnesium uptake in yeast by Alr1 requires part of the region between amino acids 240 and 322, and is not achieved by the region of Alr1 that encompasses the CorA domain. This region contains a block of charged amino acids (corresponding to amino acids 258–296 in Alr1) that is conserved between many of the fungal homologues, but is not found in the bacterial genes. It is possible that this region plays a functional role in magnesium uptake. However, the front half of the N-terminal extension of Alr1 is not essential for Mg2+ uptake, and may be serving some other function.
Deleting the C-terminal 53 amino acids of Alr1 suggested that this region is also not essential for the Mg2+ transport function of the protein. However, a frame-shift deletion mutant showed much better Mg2+ uptake than a C-terminal mutant tagged with the HA epitope. The addition of 42 amino acids containing 3x HA epitopes (YPYDVPDYA) might lead to structural distortion of the rest of the protein, and thereby affect uptake of Mg2+.
A region of Alr1 containing three putative TM domains is critical for Mg2+ transport
Random PCR mutagenesis of the 3′ half of ALR1 identified a region between amino acids 716 and 789 as being crucial for the function of Mg2+ uptake. All mutants that totally abolished uptake mapped to this critical region. Within this region are two putative TM domains predicted by computer algorithm, one of which contains the highly conserved GMN motif that is the signature of the CorA family of Mg2+ transport genes. The critical region also contains a segment immediately upstream that is homologous to a third TM domain, shown experimentally to be present in the S. typhimurium CorA protein (Smith et al. 1993). By analogy with CorA, we refer to these regions of Alr1 as putative TM1, TM2 and TM3. Of ten inactivating amino acid changes, three fell in the region of putative TM1, four within TM2, two in TM3, with the tenth (S771P) on the short loop between TM2 and TM3. One of the inactive mutants was changed in the conserved GMN motif; any changes to this motif in CorA inactivated uptake (Szegedy and Maguire 1999).
These results are consistent with the idea that the Alr1 protein might have three TM domains and so may have a topology similar to CorA (Smith et al. 1993). However, the three mutations in the putative TM1 region that completely inactivated Alr1 uptake were all non-conservative substitutions, which are expected to result in relatively gross changes of structure (see below). A domain equivalent to TM1 is not present in Mrs2 (Bui et al. 1999) or in a variant form of CorA called ZntB (Caldwell and Smith 2003). Based on the proportion of charged amino acids, it has also been suggested to be missing in a number of other CorA homologues (Smith et al. 1998; Kehres and Maguire 2002). Thus it clearly remains possible that this region of Alr1 is critical for Mg2+ transport because it forms a critical structure other than a TM domain. Direct analysis of the Alr1 protein topology is needed to determine unambiguously whether it has two or three TM domains.
Nature of the mutations
Figure 7 shows an alignment of the three TM domains of the bacterial CorA gene with the homologous regions of Alr1. Outside of the conserved GMN motif, the degree of similarity within the TM domains is not high. Alr1 amino acid residues mutated here, as well as critical residues within TM2 and TM3 of CorA (Smith et al. 1998; Szegedy and Maguire 1999) are shown in boldface.
Five of the random changes that inactivated Mg2+ transport by Alr1 involved changing amino acid residues into proline, either in putative TM1 (L725P, S729P), in TM3 (L783P, L789P) or in the loop between TM2 and TM3 (S771P). Introducing a proline is known to disrupt the structure of alpha helices (Smith and Pease 1980; Ezquerra et al. 1999), which are often important for the function of TM domains. Although it cannot be excluded that the specific residues changed to proline are essential for Mg2+ uptake, it seems likely that the severity of the phenotype associated with the proline substitutions reflects the functional importance of alpha helical structure in these critical regions.
The other five mutated residues in the “no activity” class of mutants were located in either the putative TM1 domain (R716G) or TM2 (I746V, N754L, G758C, M762L). Three of these inactivating mutations have relatively radical changes, but two of the TM2 mutations involve very conservative amino acid substitutions: I746V and M762L. We suggest that residues Ile746 and Met762 within TM2 are both critical for Mg2+ uptake. In the case of Met762, this residue is part of the highly conserved GMN motif, which has already been shown to be essential for CorA activity (Szegedy and Maguire 1999). The GMN motif has been suggested to play a role in maintaining a proper loop conformation between TM2 and TM3 rather than binding to Mg2+ (Szegedy and Maguire 1999; Kehres and Maguire 2002). In the case of Ile746, the inactivating mutation to valine provides the first indication that this residue may be critical for Mg2+ transport. Ile746 is predicted to be located close to the cytoplasmic side of the TM domain. The complete loss of function as a result of a relatively minor switch---basically the loss of a methyl group---is consistent with the idea that this amino acid plays an important role. However, unlike the GMN motif, this region of the protein and the Ile residue in particular, are not well conserved. The homologous proteins in yeast (Alr2 and Mnr2) both have a leucine at this site (MacDiarmid and Gardner 1998), while the ‘equivalent’ residue in the aligned Salmonella CorA is in fact valine (Fig. 7). Across the bacterial family, V and I are most common at this site but M, F, T, A, W and Y residues are present in different genes (Kehres et al. 1998). This valine residue was not mutated by Szegedy and Maguire (1999).

Alignment of the TM domains of the S. cerevisiae Alr1 and the S. typhimurium CorA proteins. Bold amino acids represent crucial residues experimentally identified for the putative TM domains of either Alr1 (this work) or CorA (Smith et al. 1998; Szegedy and Maguire 1999); changes to these amino acids reduced transport activity. The mutational changes described here are shown by letters above the line; bolding of the mutants indicates “no activity” mutants. The TM domains of Alr1 were predicted by alignment with CorA. The consensus (cons) residues for bacterial CorA sequences are derived from the alignments of Kehres et al. (1998)
Of the five inactivating mutations that did not involve a change to proline, four were within TM2; assuming an equal distribution of mutations during the PCR, this result suggests that the TM2 domain may be the most critical for magnesium transport. We note that one additional conservative change within TM2, V755I, resulted in a minor reduction in activity.
Two of the mutants involved independent changes to Ser729 in putative TM1; one showed no activity (S729P) and another had low activity (S729T). The latter change is a relatively conservative mutation that exchanges one hydroxyl containing side-chain for another. We therefore suggest that Ser729 in putative TM1 may also be an important residue for Mg2+ transport.
Our results also suggest that the short loop between TM2 and TM3 is important for Mg2+ transport in Alr1. The predicted size of this loop is slightly different between CorA (9 residues) and Alr1 (12 residues). Two single amino acid mutants of Alr1 were obtained in this region: S771P showed no activity, and A774S showed moderate activity. However, both mutants were reasonably radical changes. At this stage we cannot distinguish whether this short loop region plays a direct role in transport or whether it plays an indirect role in maintaining the correct location and orientation of the two adjacent TM domains, as suggested for CorA by Szegedy and Maguire (1999).
Mutagenesis of CorA indicated that hydroxyl-bearing residues in TM2 and TM3 were particularly important for Mg2+ transport (Smith et al. 1998; Szegedy and Maguire 1999). Omitting changes that involved the introduction of a proline, we obtained three mutants that involved either the introduction (M745T in TM2, A774S in the TM2-3 loop) or loss (T719A on putative TM1) of hydroxyl residues; both the TM changes were in the “low activity” class, with that in the TM2-TM3 loop showing moderate activity. Given the high number of hydroxyl-bearing residues within each TM domain (six, three and two), this is not a particularly high hit rate. However, PCR-based mutagenesis is not completely random (Fromant et al. 1995). Site-directed mutagenesis of particular residues is needed to determine whether hydroxyl residues are also important in Alr1 function.
Given the high charge density of Mg2+ ions, it might be predicted that Mg2+ transport proteins would contain negatively charged residues in the pore. However, both CorA and Alr1 completely lack charged residues on TM2 and TM3. One mutation (G790D) introduced a charge in TM3, and showed a minor reduction of Mg2+ uptake activity (but also reduced cross-reacting protein, Fig. 5). The loss of the glycine residue, rather than the introduction of a charge, may have been the important change in this instance. There are three charged residues in putative TM1 of Alr1 (two in CorA). One was mutated (R716G, Fig. 7) and gave a protein with no activity; however again this is a radical change involving glycine, so that the role of the change in charge is again not clear.
During the initial mutagenesis, some mutants with small reductions in activity were located upstream of putative TM1 within the CorA homologous region of Alr1; single mutants in this category included L495P, H541R and R620G (data not shown). Combined with the deletion analysis, these mutants provide the first evidence that specific residues in the N-terminal domain of CorA proteins are important for transport. Two further mutants were obtained immediately upstream of putative TM1: G694C (moderate activity) and L701F (low activity). Both are radical changes and might have an indirect effect by affecting the structure or location of putative TM1.
The active form of Alr1 may be a homo-oligomer
Four inactive mutants of Alr1 showed a dominant negative phenotype; that is, high-level expression of these inactive forms from a 2 μm plasmid greatly reduced the activity of wild-type Alr1 protein expressed from its chromosomal copy. Yerushalmi et al. (1996) used similar dominant negative effects to infer an oligomeric structure for EmrE, a 4-TM transport protein from E. coli. Dominant negative interactions between both homo- and hetero-oligomers have also been observed for combinations of ammonia transporters in S. cerevisiae (Marini et al. 2000) and in A. nidulans (Monahan et al. 2002).
These results are consistent with the idea that Alr1 protein forms an oligomeric complex for transport, and that oligomers containing both active and inactive proteins are themselves inactive. The results for the pCEN vector suggest that a relatively high ratio of inactive to active Alr1 may be needed for a strong negative effect.
ALR1 has been expressed in oocytes where it produced a Mg-dependent current (S. Salih, P. Donaldson and R. Gardner, unpublished data). This suggests that the protein is capable of assembling into an active transport complex without the need for any additional yeast proteins. It is possible that Alr1 may form heterodimers with Alr2, a closely related protein in yeast that is also capable of Mg2+ uptake. However, heterodimer formation is clearly not essential for transporter function, since expression of either Alr1 or Alr2 alone complemented the alr1alr2 double mutant. Thus the current evidence suggests that the most likely active structure for Alr1 is a homo-oligomer.
Structural analysis of other transport proteins with two or three TM domains have shown that they also form homotrimers (Ctr3, hCtr1) (Pena et al. 2000; Lee et al. 2002), homotetramers (KcsA, KirBac1.1) (Doyle et al. 1998; Kuo et al. 2003) or homopentamers (MscL, Chang et al. 1998). Two other members of the CorA family are known to form homo-oligomers: CorA, with three TM domains forms homotetramers (Warren et al. 2004) and Mrs2, with 2 TM domains forms homopentamers (Bui et al. 1999). In CorA, the TM domains seem to be important for homo-oligomerization. Although the N-terminal periplasmic domain of CorA retains the capability to form a homotetramer it is relatively weak in the absence of the C-terminal TM domains (Warren et al. 2004).
Conclusions
These mutagenesis results firmly establish ALR1 as a magnesium transport gene belonging to the CorA family. In particular, we have shown that the region containing the putative TM domains, including the conserved GMN motif, are essential for Mg2+ transport by Alr1. Like other characterised members of the family, it may function as a homo-oligomer. The functional role of the N-terminal and C-terminal extensions that are present only in the fungal homologues of ALR1 remains an important unanswered question. Based on conservative inactivating mutations, we have identified two new residues as functionally important for magnesium uptake: Ser729 and Ile746. Additional site-directed mutagenesis of these and other residues will help resolve their role. However, there remains an urgent need for detailed structural and electrophysiological analysis of at least one member of the CorA family in order to place these data in an appropriate context.
Abbreviations
- TM:
-
transmembrane
- YPD:
-
yeast extract peptone dextrose
- YPDM:
-
YPD plus high magnesium
- LPM:
-
low pH low magnesium
- SC:
-
synthetic complete
- OD:
-
Optical density
- HA:
-
Haemaglutenin
- ER:
-
endoplasmic reticulum
- PCR:
-
Polymerase chain reaction
References
Bonneaud N, Ozierkalogeropoulos O, Li GY, Labouesse M, Minviellesebastia L, Lacroute F (1991) A family of low and high copy replicative, integrative and single-stranded Saccharomyces-cerevisiae Escherichia-coli shuttle vectors. Yeast 7:609–615
Bui DM, Gregan J, Jarosch E, Ragnini A, Schweyen RJ (1999) The bacterial magnesium transporter CorA can functionally substitute for its putative homologue Mrs2p in the yeast inner mitochondrial membrane. J Biol Chem 274:20438–20443
Caldwell AM, Smith RL (2003) Membrane topology of the ZntB efflux system of Salmonella enterica Serovar Typhimurium. J Bacteriol 185:374–376
Casagrande R, Stern P, Diehn M, Shamu C, Osario M, Zuniga M, Brown PO, Ploegh H (2000) Degradation of proteins from the ER of S. cerevisiae requires an intact unfolded protein response pathway. Mol Cell 5:729–735
Chang G, Spencer RH, Lee AT, Barclay MT, Rees DC (1998) Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science 282:2220–2226
Doyle DA, Cabral JaoM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280:69–77
Drummond RSM, Tutone A, Li Y-C, Gardner RC (2005) A putative magnesium transporter AtMRS2–11 is localized to the chloroplastic envelope membrane. Plant Sci (in press)
Ezaki B, Sivaguru M, Ezaki Y, Matsumoto H, Gardner RC (1999) Acquisition of aluminum tolerance in Saccharomyces cerevisiae by expression of the BCB or NtGDI1 gene derived from plants. FEMS Microbiol Lett 171:81–87
Ezquerra M, Carnero C, Blesa R, Gelpi JL, Ballesta F, Oliva R (1999) A presenilin 1 mutation (Ser169Pro) associated with early-onset AD and myoclonic seizures. Neurology 52:566–570
Fromant M, Blanquet S, Plateau P (1995) Direct random mutagenesis of gene-sized DNA fragments using polymerase chain-reaction. Anal Biochem 224:347–353
Fuhrmann GF, Rothstein A (1968) The transport of Zn2+, Co2+ and Ni2+ into yeast cells. Biochim Biophys Acta 163:325–330
Gardner RC (2003) Genes for magnesium transport. Curr Opin Plant Biol 6:263–267
Gietz RD, Schiestl RH, Willems AR, Woods RA (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11:355–360
Graschopf A, Stadler JA, Hoellerer MK, Eder S, Sieghardt M, Kohlwein SD, Schweyen RJ (2001) The yeast plasma membrane protein Alr1 controls Mg2+ homeostasis and is subject to Mg2+-dependent control of its synthesis and degradation. J Biol Chem 276:16216–16222
Grubbs RD, Snavely MD, Paul Hmiel S, Maguire ME (1989) [36] Magnesium transport in eukaryotic and prokaryotic cells using magnesium-28 ion. Methods in enzymology, vol 173. Academic, New York, pp 546–563
Hmiel SP, Snavely MD, Miller CG, Maguire ME (1986) Magnesium transport in Salmonella typhimurium: characterization of magnesium influx and cloning of a transport gene. J Bacteriol 168:1444–1450
Kehres DG, Lawyer CH, Maguire ME (1998) The CorA magnesium transporter gene family. Microb Comp Genomics 3:151–169
Kehres DG, Maguire ME (2002) Structure, properties and regulation of magnesium transport proteins. BioMetals 15:261–270
Kolisek M, Zsurka G, Samaj J, Weghuber J, Schweyen RJ, Schweigel M (2003) Mrs2p is an essential component of the major electrophoretic Mg2+ influx system in mitochondria. EMBO J 22:1235–1244
Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA (2003) Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300:1922–1926
Kushnirov VV (2000) Rapid and reliable protein extraction from yeast. Yeast 16:857–860
Lee J, Pena MMO, Nose Y, Thiele DJ (2002) Biochemical characterization of the human copper transporter Ctr1. J Biol Chem 277:4380–4387
Lee K, Neigeborn L, Kaufman RJ (2003) The unfolded protein response is required for haploid tolerance in yeast. J Biol Chem 278:11818–11827
Li L, Tutone AF, Drummond RS, Gardner RC, Luan S (2001) A novel family of magnesium transport genes in Arabidopsis. Plant Cell 13:2761–2775
Liu GJ, Martin DK, Gardner RC, Ryan PR (2002) Large Mg(2+)-dependent currents are associated with the increased expression of ALR1 in Saccharomyces cerevisiae. FEMS Microbiol Lett 213:231–237
Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961
MacDiarmid CW, Gardner RC (1996) A1 toxicity in yeast. A role for Mg? Plant Physiol 112:1101–1109
MacDiarmid CW, Gardner RC (1998) Overexpression of the Saccharomyces cerevisiae magnesium transport system confers resistance to aluminum ion. J Biol Chem 273:1727–1732
Marini AM, Springael JY, Frommer WB, Andre B (2000) Cross-talk between ammonium transporters in yeast and interference by the soybean SAT1 protein. Mol Microbiol 35:378–385
Monahan BJ, Unkles SE, Tsing IT, Kinghorn JR, Hynes MJ, Davis MA (2002) Mutation and functional analysis of the Aspergillus nidulans ammonium permease MeaA and evidence for interaction with itself and MepA. Fungal Genet Biol 36:35–46
Muhlrad D, Hunter R, Parker R (1992) A rapid method for localized mutagenesis of yeast genes. Yeast 8:79–82
Patil C, Walter P (2001) Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol 13:349–355
Pena MMO, Puig S, Thiele DJ (2000) Characterization of the Saccharomyces cerevisiae high affinity copper transporter Ctr3. J Biol Chem 275:33244–33251
Schock I, Gregan J, Steinhauser S, Schweyen R, Brennicke A, Knoop V (2000) A member of a novel Arabidopsis thaliana gene family of candidate Mg2+ ion transporters complements a yeast mitochondrial group II intron-splicing mutant. Plant J 24:489–501
Serrano R, Monk BC, Villalba JM, Montesinos C, Weiler EW (1993) Epitope mapping and accessibility of immunodominant regions of yeast plasma membrane H(+)-ATPase. Eur J Biochem 212:737–744
Sherman F (1991) Getting started with yeast. Methods Enzymol 194:3–21
Smith JA, Pease LG (1980) Reverse turns in peptides and proteins. Crc Crit Rev Biochem 8:315–399
Smith RL, Banks JL, Snavely MD, Maguire ME (1993) Sequence and topology of the CorA magnesium transport systems of Salmonella typhimurium and Escherichia coli. Identification of a new class of transport protein. J Biol Chem 268:14071–14080
Smith RL, Szegedy MA, Kucharski LM, Walker C, Wiet RM, Redpath A, Kaczmarek MT, Maguire ME (1998) The CorA Mg2+ transport protein of Salmonella typhimurium. Mutagenesis of conserved residues in the third membrane domain identifies a Mg2+ pore. J Biol Chem 273:28663–28669
Svetlov V, Cooper TG (1998) Efficient PCR-based random mutagenesis of sub-genic (100 bp) DNA fragments. Yeast 14:89–91
Szegedy MA, Maguire ME (1999) The CorA Mg(2+) transport protein of Salmonella typhimurium. Mutagenesis of conserved residues in the second membrane domain. J Biol Chem 274:36973–36979
Warren MA, Kucharski LM, Veenstra A, Shi L, Grulich PF, Maguire ME (2004) The CorA Mg2+ transporter is a homotetramer. J Bacteriol 186:4605–4612
Worlock AJ, Smith RL (2002) ZntB is a novel Zn2+ transporter in Salmonella enterica serovar Typhimurium. J Bacteriol 184:4369–4373
Yerushalmi H, Lebendiker M, Schuldiner S (1996) Negative dominance studies demonstrate the oligomeric structure of EmrE, a multidrug antiporter from Escherichia coli. J Biol Chem 271:31044–31048
Zsurka G, Gregan J, Schweyen RJ (2001) The human mitochondrial Mrs2 protein functionally substitutes for its yeast homologue, a candidate magnesium transporter. Genomics 72:158–168
Acknowledgements
We thank Keith Richards for technical assistance, Mark Longtine (Oklahoma) for supplying plasmids and Ramon Serrano (Valencia) for supplying PMA1 antibody, and Colin MacDiarmid and Alok Mitra for reviewing versions of the manuscript. J-M L. was the recipient of an Overseas Scholarship from the Korean government during his PhD. Funding for this project was provided by a grant from the New Zealand Marsden Fund.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann
Rights and permissions
About this article
Cite this article
Lee, Jm., Gardner, R.C. Residues of the yeast ALR1 protein that are critical for Magnesium uptake. Curr Genet 49, 7–20 (2006). https://doi.org/10.1007/s00294-005-0037-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-005-0037-y