
Abstract
ATM/ATR homologs are the central elements of genome surveillance mechanisms in many organisms, including yeasts, flies, and mammals. In Saccharomyces cerevisiae, most checkpoint responses depend on the ATR ortholog Mec1p. The yeast ATM ortholog, Tel1p, so far has been implicated in a specific DNA damage checkpoint during S-phase as well as in telomere homeostasis. In particular, yeast cells lacking only Tel1p harbor short but stable telomeres, while cells lacking both Tel1p and Mec1p are unable to maintain telomeric repeats and senesce. Here, we present the characterization of a new mutation in the TEL1-gene, called tel1-11, which was isolated by virtue of a synthetic lethal interaction at 37°C with a previously described mec1-ts mutation. Interestingly, telomere and checkpoint functions are differentially affected by the mutant protein Tel1-11p. The Tel1p-dependent checkpoint response is undetectable in cells containing Tel1-11p and incubated at 37°C, but basic telomere function is maintained. Further, when the same cells are incubated at 26°C, Tel1-11p confers full proficiency for all telomere functions analyzed, whereas the function for DNA-damage checkpoint activation is clearly affected. The results thus strongly suggest that the different cellular pathways affected by Tel1p do not require the same level of Tel1p activity to be fully functional.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In eukaryotes, maintaining genome integrity relies on a set of surveillance mechanisms, called checkpoints. These checkpoints are responsible for proper detection and repair of DNA damage caused by environmental stresses or irregularities during DNA replication. Pivotal elements of all known eukaryotic checkpoints are homologs of mammalian ATM/ATR kinases (ataxia-telangiectasia-mutated/ATM and Rad3-related (Nyberg et al. 2002). In budding yeast, Mec1p (an ATR ortholog), plays a prevalent role in G1, S, and G2/M checkpoints. In addition to sensitivity to all DNA-damaging agents, Mec1p-deficient cells display increased gross chromosomal rearrangements (GCR) and aberrant patterns of DNA replication (Santocanale and Diffley 1998; Myung et al. 2001; Cha and Kleckner 2002). A downstream target of Mec1p-dependent phosphorylation is another essential kinase, Rad53p (Sanchez et al. 1996). The essential function of Mec1p and Rad53p is thought to be up-regulation of nucleotide pools in response to replication and DNA damage, as alleviation of inhibition of nucleotide synthesis in sml1 mutants rescues the lethality of cells lacking Mec1p or Rad53p (Zhao et al. 1998).
The checkpoint function of Tel1p, the budding yeast ortholog of ATM, appears more furtive, as Tel1p-deficient cells do not suffer from obvious sensitivity to DNA-damaging agents and there is no major defect in genome stability, as assessed by GCR rates (Morrow et al. 1995; Myung et al. 2001). In addition, Telp1 appears not to be involved in the induction of nucleotide synthesis, at least after DNA damage (Zhu and Xiao 2001). Tel1p’s contribution to cellular checkpoint activity might be masked by the prevailing activity of Mec1p. For example, a Tel1p deficiency exacerbates checkpoint defects and causes large increases of GCR rates displayed by mec1 mutants (Morrow et al. 1995; Myung et al. 2001). Furthermore, in response to DNA damage, Rad53p can be phosphorylated in both Mec1p and Tel1p-dependent manner, although Tel1p’s dependent phosphorylation is minor and is detectable mainly in cells lacking Mec1p (Sanchez et al. 1996; Vialard et al. 1998; Clerici et al. 2001; Nakada et al. 2003b).
According to a current view, most DNA damage is converted to DNA intermediates with single-stranded DNA (ssDNA). These intermediates are readily sensed by ATR-dependent pathways, whereas the ATM-dependent pathway is thought to respond to DNA double-strand breaks (DSB). By extension, this might explain why the checkpoint function provided by Mec1p prevails over that of Tel1p (Nyberg et al. 2002). Supporting this view, a striking demonstration of a Tel1p-dependent checkpoint function could be made in yeast cells in which conversion of DSB into ssDNA intermediates is blocked by sae2 or rad50S mutations. In mitotic cells lacking both Mec1p and Sae2p, the sensitivity to methyl methanesulfonate (MMS) is partially suppressed and such cells are capable of Rad53p phosphorylation and cell cycle arrest. It was shown that these checkpoint functions are dependent on Tel1p and Mre11p (also referred to as Tel1p- and Mre11p-dependent checkpoint or TM-checkpoint; (Usui et al. 2001). However, Mre11p has multiple implications in G1 and G2/M checkpoints (D’Amours and Jackson 2002) and it remains unclear how specific its implication in a S-phase DNA-damage checkpoint is. In yeast, Mre11p forms a complex with Rad50p and Xrs2p, called the MRX-complex. Interestingly, under particular conditions and in response to DNA damage, both Mre11p and Xrs2p have been reported to be phosphorylated in a Tel1p-dependent fashion (D’Amours and Jackson 2001; Usui et al. 2001; Nakada et al. 2003b).
An additional challenge for DNA replication and therefore genomic stability is the faithful maintenance of chromosomal termini, the telomeres. As could be predicted, subsets of proteins involved in checkpoint pathways have a role to play in telomere maintenance. Indeed, certain mutations in MEC1 or RAD53 cause a shortening of telomeric repeat tracts and a decrease in telomere position effect (TPE, telomeric chromatin-dependent transcriptional inhibition) (McAinsh et al. 1999; Ritchie et al. 1999). Furthermore, mutations in genes of the Rad17-Mec3-Ddc1 complex, which is involved in the Mec1p-dependent pathway, can also affect telomere length (Corda et al. 1999; Longhese et al. 2000); Dionne and Wellinger, unpublished data). Furthermore, Mec1p has been shown to associate with telomeres during late S phase (Takata et al. 2004). More recent data on the assembly of repair complexes at DSB as well as studies on the association of proteins at telomeres suggest that the MRX-complex and Telp1p are among the first factors to associate to such DNA-ends (Lisby et al. 2004; Takata et al. 2005). Taken together, while a deficiency in the Mec1p-dependent pathway clearly has some influence at telomeres, no telomere-specific function could be assigned to its components yet.
In contrast, both Tel1p and its mammalian ortholog ATM have been directly implicated in telomere homeostasis (Pandita 2002). Both in mice and human cells, an ATM deficiency provokes accelerated telomere loss and other phenotypic changes such as genomic instability (Metcalfe et al. 1996; Wong et al. 2003). In yeast, the TEL1 gene had initially been identified in a screen for mutations affecting telomere length and Tel1p-deficient cells have very short telomeric repeat tracts (Lustig and Petes 1986). Later genetic analyses placed TEL1 and the genes encoding the MRX-complex) in the same pathway as telomerase (Ritchie et al. 1999; Ritchie and Petes 2000). The same analyses showed that in the absence of these proteins, cells rely on Mec1p to maintain short but stable telomeres, which suggests that the function of Mec1p at telomeres is redundant with or masked by the Tel1p and the MRX-complex in normal cells. In the complete absence of ATM-like kinases, yeast cells are not able to maintain telomeric DNA and senesce after about 60–80 generations, a phenotype also displayed by cells lacking telomerase components (Ritchie et al. 1999; Chan et al. 2001). One characteristic of this senescence phenotype is the emergence of survivors: these are cells that can maintain telomeric DNA in a telomerase-independent fashion but relying on homeologous recombination (Lundblad and Blackburn 1993). Finally, telomerase and Tel1p have recently been associated with a chromosome protective function that may be independent of active telomere lengthening (Chan and Blackburn 2003). Intriguingly, the two ATM-like kinases, Mec1p and Tel1p, show mutually exclusive association with telomeres at distinct stages of the cell cycle (Takata et al. 2004). However, the molecular mechanisms for Mec1p- or Tel1p-dependent telomere dynamics remain unclear and hypotheses are dependent on model systems and conditions used (Takata et al. 2004); reviewed in (Chakhparonian and Wellinger 2003). It would therefore be of interest to be able to analyze mutant Tel1p proteins that cause deficiencies in only one pathway, but not for others. There is a precedence suggesting that the functions of proteins involved in DNA-damage checkpoint and in telomere homeostasis may be separable. Cells expressing a particular mutant Mre11p protein, called Mre11-tsp, display short telomeres, similar to those observed in cells lacking Mre11p altogether (Chamankhah et al. 2000). However, these cells are not sensitive to an exposure to MMS when grown at 24°C, while they are sensitive to MMS at 34°C. These data show that the various functions of Mre11p can be affected in a differential fashion and perhaps separable.
Here we describe the characterization of a mutation in the TEL1 gene, generating a protein we call Tel1-11p. Cells harboring this protein display properties that are consistent with the hypothesis that the functions of Tel1p at telomeres and in the DNA-damage checkpoint have different requirements for Tel1p function. We analyzed three parameters to characterize the effects of the Tel1-11p on cells. Checkpoint functionality was assessed as resistance to MMS-exposure and induction of Rad53p-phosphorylation in cells that contained the Tel1-11p in combination with mec1Δsml1Δsae2Δ mutations. Telomere functions were assessed as overall telomere length and, independently, the ability to stably maintain a certain telomeric repeat tract when Tel1-11p expression is combined with mec1Δsml1Δ mutations. The results show that when the cells are grown at 26°C, Tel1-11p confers completely wild-type telomere homeostasis and its checkpoint function is slightly affected. Upon growth of the cells at 37°C, telomeres are shortened, but still stably maintained, even in the absence of Mec1p. However, the checkpoint function is severely impaired or absent at this temperature, since after exposure of cells to MMS, Rad53p phosphorylation is undetectable. These data thus strongly suggest a differential requirement for Tel1p activity in telomere maintenance as compared to the checkpoint function.
Materials and methods
Strains and plasmids
Saccharomyces cerevisiae strains used in this study are listed in Table 1. All yeast cells were grown at temperatures indicated in either complete YPD supplemented at need with nourseothricin at 100 μg/ml (WERNER Bioagents, Germany) or G-418 at 150 μg/ml (Sigma). Alternatively, synthetic medium YC supplemented with required amino acids and bases was used. Genetic manipulations were as described (Zakian and Scott 1982; Rose et al. 1990). Transformations were performed according to a modified lithium acetate method (Gietz et al. 1995).
Haploid strains were produced by micro-dissection and genotypes confirmed by marker segregation and Southern analyses, if needed. For construction of the diploid RWY51 (Table 1), the mec1-ts allele (Weinert et al. 1994) was recreated in diploid BY4705 (Brachmann et al. 1998) by integrating a copy of mec1-ts linked to the HIS3 gene into the MEC1 locus, creating mec1::mec1-ts::HIS3 (T. Weinert, unpublished data). Subsequently, the tel1Δ::LEU2 and sml1Δ::TRP1 deletions were introduced into that strain by successively replacing the open reading frames (ORF) of the corresponding genes with the respective selection markers using a PCR-mediated gene disruption method (Brachmann et al. 1998). The same technique was used to introduce the sae2Δ::kanMX4 and rad52Δ::kanMX4 deletions into strains as indicated in Table 1. E. coli strain DH5α was used for plasmid manipulation according to standard procedures (Sambrook et al. 1989).
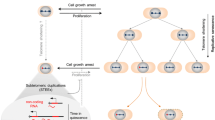
Screen for mutations conferring conditional synthetic lethality with mec1-ts (see Fig. 1 for a general outline)
RWY51-24A cells (mec1-ts ade2 ura3) carrying a p[MEC1, ADE2, URA3] plasmid were subjected to EMS-induced mutagenesis. Briefly, exponentially growing cells were collected, washed twice and resuspended in 1.5 ml of 0.01 M potassium phosphate buffer at pH 7.0, sonicated lightly and incubated for 30 min in presence of 2% EMS, leading to about 40–50% lethality. They were then washed thrice in water, plated on YC medium containing 10 mg/l adenine and incubated at 37°C. An aliquot of cells was also incubated at 26°C as a scoring control. Cell growth at 37°C was observed up to 7 days after plating and white non-sectored colonies were selected as clones dependent on the plasmid. About 50,000 colonies were screened in this manner. In order to purify the background and select clones that were affected at a single locus, candidate strains were backcrossed to wild type three times. Since these backcrosses involved at least two loci (the mec1-ts allele and the unknown mutation, xxx), haploid segregants after microdissection were expected to follow the segregation ratio of 4:1:1 (tetratype:parental ditype:non-parental ditype). For example, for the strain leading to the eventual identification of the tel1-11 allele, 34 tetrads score yielded 24:6:4 (T:PD:NPD) tetrads. Furthermore, when a mec1-ts tel1-11 strain was crossed to mec1-ts strain, haploid segregants after microdissection followed the segregation ratio of 2:2 (mec1-ts phenotype:mec1-ts tel1-11 phenotype) for 29 of 31 tetrads analyzed, confirming a single mutation unlinked to MEC1. Next, we assessed whether the selected clones contained a mutation in any of the genes known to display synthetic lethality with mec1-ts at 37°C, namely TEL1, MRE11, XRS2, RAD50, or RAD52. This was achieved by (1) crossing candidate double mutants (mec1-ts, xxx) with respective double mutants and checking the viability of diploids at 37°C; and (2) by complementing haploid double mutants (mec1-ts, xxx) with plasmids carrying one of the genes above (Fig. 1a). These experiments identified at least one mutation in RAD50 and two in TEL1. Haploid cells harboring the mutation in RAD50, or one of the mutations in TEL1, together with the mec1-ts allele displayed short telomeres when grown at 26°C, as expected, whereas one of the TEL1 alleles, named tel1-11, conferred wt telomere lengths (Fig. 1b). This latter allele was isolated and re-introduced into the genome as described below.

Schematic presentation of the conditional synthetic lethality screen that allowed identification of the tel1-11 allele. a Experimental chart flow. xxx represents a mutant allele of candidate genes conferring synthetic lethality at 37°C if combined with the mec1-ts allele. b Cells with the indicated genotypes were grown for at least 90 generations at 26°C on plates and then allowed to grow for an additional five generations in liquid culture prior to DNA extraction and TRF analyses. Lane 1 RWY51-23A (mec1-ts tel1Δ); lane 2 RWY51-24A (mec1-ts TEL1); lane 3 clone 11 (mec1-ts tel1-11); lane 4 clone 5 (mec1-ts rad50)
Allele rescue and sequencing
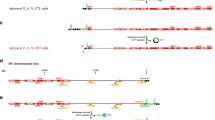
See Fig. 2a for a general outline of the TEL1 locus. Plasmid p316TGR contains the TEL1 gene region fragments −4,582 to −4,159 and −388 to +9,133 with respect to the translation initiation codon. These regions were connected by a polylinker containing a SacII recognition site and the fragment was inserted into the SacI–SalI sites of pRS316 (Sikorski and Hieter 1989). For allele rescue, p316TGR was digested with SacII and NheI to create a gap in TEL1 from −4,159 to +8,830 and the gapped plasmid was transformed into yeast strains carrying either TEL1 or the tel1-11 alleles. The plasmids resulting from this allele rescue (Fig. 2b) were named p316GRtel1-11 and p316GRTEL1, respectively. The complete tel1-11 coding region on the p316GRtel1-11 plasmid was sequenced at least twice using internal TEL1 primers and the assembled sequence revealed a single point mutation (G3955A), leading to a E to K change at amino acid position 1319. Further, a SpeI-fragment spanning the region from −808 to +6,300 was subcloned and sequenced using flanking T3/T7 and internal TEL1 primers, revealing no additional mutations in this entire region. In order to be able to follow TEL1-alleles through crosses, plasmids p316GRtel1-11natR and p316GRTEL1natR were constructed by inserting a PCR-amplified natR selection marker (conferring resistance to nourseothrecin (Goldstein and McCusker 1999) into the BstXI sites upstream of the TEL1 gene of p316GRtel1-11 and p316GRTEL1 plasmids, respectively (Fig. 2c). Integration of the natR-linked tel1-11 or TEL1 alleles into the genome was performed by transformation of the RWY51-23A strain with PmlI- SalI fragments (Fig. 2c). Replacement of tel1Δ::LEU2 by tel1-11::natR, TEL1::natR or tel1Δ::LEU2::natR was verified by Southern blotting and phenotype analysis of the corresponding strains (data not shown). Strains with or without the natR gene integrated upstream from different alleles of TEL1 gene behaved similarly in all assays used (data not shown). Plasmid p316TEL1 contained a TEL1 genomic fragment −808 to ∼+9,133 in the SpeI–SalI sites of pRS316 and was used interchangeably with p316GRTEL1 in complementation assays.

Organization of the TEL1 locus. a TEL1 genomic region; b Fragments recovered by gap repair from TEL1 or tel-11 carrying strains (mutation in tel1-11 is indicated); c Introduction of tel-11::natR or TEL1::natR into the genome. Featured are TEL1 ORF (gray box), domains: FAT (vertically striped box), kinase (dotted box) and FATC (horizontally striped box) (Bosotti et al. 2000). Marker genes [LEU2, natR (black box)] and flanking fragments used for gap repair (hatched boxes) are also indicated. Vertical bars represent: G3955A—the position of the mutation revealed by sequencing, BstXI—the sites delimiting the region that was replaced by natR; PmlI, SacII and NheI—sites used for gap repair or genomic integration of TEL1 alleles. The SacII site is indicated in brackets as it does not belong to the genomic sequence but only to the p316TGR plasmid; it consequently disappeared during the gap repair
Yeast senescence assays and growth tests
Senescence was assessed by visual analysis of colony-formation and growth on solid-rich medium (YPD) under given conditions for a number of generations, and further confirmed by telomere analysis (see below; Lundblad and Szostak 1989). Formation of an average colony from a single cell was estimated to require ∼20 divisions, and growth in a 5 ml liquid culture to saturation ∼5 generations. Overall ability to grow was evaluated by “spot dilution tests”. The cell concentration of an initial liquid culture was determined by measuring the OD600 and corresponding numbers of cells were spotted in serial tenfold dilutions on YPD medium.
MMS sensitivity assays
Stationary cultures were diluted to ∼0.5–1.5×106 cells/ml and allowed to re-grow to exponential phase (∼0.5–1.5×107 cells/ml). For permanent exposure to MMS, cell growth was assessed by spot dilution tests on YPD medium containing indicated concentrations of MMS. Scoring was generally performed after a 3 or 4-day growth before MMS concentration declined due to its degradation. For acute exposure to MMS, MMS was added directly (final concentration 0.01%) to a culture which was usually 5–20 ml. These MMS-treated cultures were incubated for 90 min at the indicated temperatures, then washed thrice with water and survival was assessed by spot dilution tests on YPD medium lacking MMS. Colony scoring was generally performed after a 3-day growth, as no distinct additional colonies appeared later.
Analysis of telomere length
Telomere length was analyzed as described (Wellinger et al. 1993). Briefly, isolated genomic DNA was digested with the XhoI restriction enzyme, which cuts once in the conserved telomere proximal Y′ repeat element. Many yeast telomeres contain such a Y′-element and this digestion releases a diagnostic ∼1.2 kb terminal restriction fragment (TRF), which includes the terminal ∼0.35 kb TG1-3 repeats. This DNA is then analyzed by Southern blotting using specific TG1-3-repeat probes, as described previously (Wellinger et al. 1993).
Western analysis
After acute exposure to MMS (see above), protein extracts from 5 to 20 ml aliquots of the cultures were prepared using a modified TCA method. The same cultures were also used to assess survival by spot dilution tests. Proteins were separated on an 8% acrylamid-bisacrylamid gel (ratio 37.5:1) according to standard techniques (Sambrook et al. 1989) and transferred onto Hybond-P membrane according to supplier’s instructions (Amersham Pharmacia Biotech). Anti-Rad53p polyclonal rabbit antibody was a kind gift from Frederic Sweeney and Daniel Durocher (Samuel Lunenfeld Research Institute, University of Toronto). Chemiluminescent detection of antigen–antibody complexes was carried out with horseradish peroxidase-conjugated anti-rabbit secondary antibodies in combination with ECL+ detection reagent (Amersham Pharmacia Biotech).
Results
Isolation of a mutation in the TEL1 gene that is lethal when combined with a mec1-ts mutation
Cells harboring the mec1-ts allele grow normally at 26°C and are only mildly impaired at 37°C (Fig. 3). They are moderately resistant to the DNA-damaging agent MMS at both 26 and 37°C, and arrest properly upon a single HO-induced DSB at 26°C, but not at 36°C (Weinert et al. 1994). Interestingly, the mec1-ts mutation confers lethality when combined with a deletion of TEL1 (Fig. 3), MRE11 or RAD50 (data not shown). This suggests that an essential function of Mec1p is provided by these genes in mec1-ts cells at 37°C. The lethality of mec1-ts tel1Δ cells at 37°C is suppressed by a deletion of SML1 or over-expression of RNR1 (data not shown), indicating that this essential function is related to regulation of nucleotide synthesis (Zhao et al. 1998). However, it is not clear to which extent the essential function of the mec1-ts-encoded protein is compromised, as the mec1-ts allele has not been characterized in full detail. We initially wished to determine which, if any, other genes would belong to the same pathway as the TEL1 and MRX-genes. Therefore, we performed a plasmid-loss based genetic screen (Kranz and Holm 1990) for mutations that would cause lethality when combined with the mec1-ts mutation at 37°C, but not at 26°C. Primary candidate mutants were identified as mec1-ts cells that could not lose a plasmid harboring a wild-type copy of MEC1 gene at 37°C, but could lose the same at 26°C (see Materials and methods). As expected, we recovered single-locus mutants that affected the RAD50 and TEL1 genes. Curiously however, one of the single recessive mutations isolated in our screen, clone 11, while genetically linked to TEL1, conferred normal TRF sizes when the cells were grown at 26°C (see below). These initial results indicated that this mutation in TEL1 did not yield a short telomere phenotype at 26°C, yet it conferred synthetic lethality when combined with the mec1-ts allele at 37°C.

At 37°C the tel1-11 mutation is lethal when combined with the mec1-ts mutation. Growth tests of integrative mutants were performed as described in Materials and methods. About ten cells were plated on the most diluted spots. Strains used are: 1 MCY56-4 (mec1-ts TEL1); 2 MCY56-10 (mec1-ts TEL1); 3 MCY511-1 (mec1-ts tel1-11); 4 MCY511-3 (mec1-ts tel1-11); 5 MCY56-3 (mec1-ts tel1Δ); 6 MCY511-9 (mec1-ts tel1Δ)
In order to ensure that the mutation indeed occurred in the TEL1 gene, the mutated gene was cloned by gap repair using a plasmid bearing a gapped copy of TEL1 (see Materials and methods). The complete region corresponding to a functional TEL1 gene was sequenced and shown to contain one single nucleotide divergence with the TEL1 wild-type sequence. The detected guanine to adenine substitution is predicted to provoke the amino acid change E1319K in the Tel1p sequence. The corresponding allele was thus named tel1-11 and the protein encoded by it Tel1-11p.
In order to further characterize this mutation, we integrated the tel1-11 into the genome of the strain RWY51-23A in replacement of tel1Δ::LEU2 as described in Materials and methods. The analysis of the resultant strain proved that tel1-11 conferred synthetic lethality with mec1-ts at 37°C, but did not impair the growth of mec1-ts cells at 26°C (Fig. 3). We then characterized the tel1-11 allele with respect to three functions that the Tel1p protein is known to accomplish: normal telomere length maintenance, maintenance of telomeres in mec1Δsml1Δ cells and activation of a DNA-damage checkpoint.
In cells expressing Tel1-11p, telomere lengths are normal at 26°C and shortened at 37°C
In order to assess the telomere functions of Tell-11p, congenic spores bearing corresponding alleles of TEL1 were derived from diploid heterozygous strains MCY751 (MEC1/mec1-ts tel1Δ/tel1-11) or RWY51 (MEC1/mec1-ts TEL1/tel1Δ). These heterozygous diploids, when grown at 26°C, possessed normal telomeres (data not shown). Haploid segregants were sub-cultured by re-streaking on plates at 26 or 37°C followed by a liquid culture at the same conditions, genomic DNA was isolated and subjected to TRF analysis. In cells harboring certain TEL1 alleles or upon a complete loss of the TEL1 gene, telomeres shorten to attain their minimal stable length after about 150 generations (Ritchie et al. 1999; Lustig and Petes 1986). After growth for 145 generations at 26°C, the presence of Tel1-11p still conferred stable telomere lengths, which were indistinguishable from cells containing the wild-type Tel1p (Fig. 4a). As reported before, strains lacking Tel1p (tel1Δ) bore short stable telomeres. When the cells were subcultured at 37°C, telomeres of tel1-11 strains gradually shortened, but after about 145 generations, they were still slightly longer than those of cells lacking Tel1p altogether (Fig. 4b, lanes 7–12). When these tel1-11 cells were shifted back to 26°C, their telomeres regained wild-type length within 45 generations (Fig. 4b, lanes 1–6). These data indicate that Tel1-11p confers some temperature sensitivity in terms of normal telomere length maintenance: this function appears to be fully supplied when cells are grown at 26°C, but compromised at 37°C.

The Tel1-11p confers a normal telomere length phenotype at 26°C and shortened telomeres at 37°C. a Cells harboring the indicated alleles of the TEL1 gene were cultivated at 26°C for 145 generations and TRF lengths were assessed as described in Materials and methods. b Cells as in (a) were cultivated for 120 generations at 37°C, then split into two subcultures; one was kept for an additional 25 generations at 37°C (lanes 7–12, right), the other was cultivated for the next 45 generations at 26°C (lanes 1–6, left). M molecular weight marker, G generations. Signals between 1.0 and 1.6 kb are TRFs from Y’ telomeres; some non-Y’ TRFs of >1.6 kb can be seen. Strains used are 1, 7 RWY51-7B (TEL1); 2, 8 RWY51-28C (TEL1); 3, 9 MCY751-1A (tel1Δ); 4, 10 MCY751-2C (tel1Δ); 5, 11 MCY751-1D (tel1-11); 6, 12 MCY751-2B (tel1-11)
Mec1Δ sml1Δ tel1-11 cells can maintain short, but stable telomeres at 37°C
As a second criterion for the functioning of Tel1-11p at telomeres, we asked whether tel1-11 cells depended on Mec1p to maintain a critical telomere length without entering a senescence crisis. To that end, we first compared the growth and telomere TRF length patterns of cells that were grown at 37°C with the following genotypes: mec1Δsml1ΔTEL1 rad52Δ, mec1Δsml1Δtel1-11 rad52Δ and mec1Δsml1Δtel1Δrad52Δ. No striking difference could be observed in the growth of Mec1-deficient cells possessing a TEL1 or tel1-11 allele for 120 generations analyzed, whereas cells with a tel1Δ allele displayed a clear senescent phenotype after ∼60 generations (Fig. 5a). Furthermore, at the end of the analyses, mec1Δsml1Δtel1-11 rad52Δ cells harbored short and stable telomeres (Fig. 5b). As a possible indication of a stable maintenance of the telomeres in this strain, they were clearly longer than those of mec1Δsml1Δtel1Δ cells and there was no indication of telomeric rearrangements typical for survivors, which maintain their telomeres by homeologous recombination (Fig. 5b, Lundblad and Blackburn 1993; Teng and Zakian 1999). These data indicate that even when cells were grown continuously at 37°C, cells lacking Mec1p and expressing Tel1-11p can stably maintain telomeric repeats and do not display phenotypes that would indicate a complete loss of telomerase activity at the telomeres, such as observed in the cells completely lacking both Mec1p and Tel1p. Thus, although Tel1-11p cannot fully supply the function to maintain normal length telomeres at 37°C, this protein is proficient in providing the functions required for stable maintenance of telomeric repeats in the absence of Mec1p at all temperatures.

In the absence of Mec1p, Tel1-11p does not confer a senescence phenotype even when the cells are grown at 37°C. a Subculturing of strains on plates were performed at 37°C for the indicated number of generations as described in Materials and methods. To the right, the relevant genotypes of the strains cultured in the respective quadrants of the plates are indicated. Note that mec1Δsml1Δtel1Δrad52Δ cells (top left quadrant) did not grow beyond 80 generations. Strains used are: MCY1700-7A (mec1Δsml1Δrad52Δtel1Δ); MCY1700-2C (mec1Δsml1Δrad52ΔTEL1); MCY111-6A, MCY111-10A (mec1Δsml1Δtel-11 rad52Δ). b Cells with the indicated genotypes were grown for at least 120 generations at 37°C on plates and were then allowed to grow for an additional five generations in liquid culture prior to DNA extraction and TRF analyses as described in Materials and methods. Lane 1 MCY1700-2C (mec1Δsml1Δrad52ΔTEL1); lane 2 MCY111-6A (mec1Δsml1Δrad52Δtel1-11); lane 3 DFY 030 (mec1Δsml1ΔRAD52 tel1Δ)
The DNA-damage checkpoint function is undetectable in cells expressing Tel1-11p grown at 37°C
In the presence of Mec1p, strains expressing Tel1-11p did not display any obvious growth defects or MMS sensitivity at either 26 or 37°C (data not shown). This was expected though, since otherwise wild-type cells but lacking Tel1p altogether were shown to behave similarly (Sanchez et al. 1996). However, the contribution of Tel1p to the a DNA-damage checkpoint was demonstrated in cells that contain mec1Δsml1Δsae2Δ mutations (Usui et al. 2001). We therefore introduced the tel1-11 allele into that background by crossings (see Table 1). In this setting, cells possessing a functional checkpoint provided by Tel1p are relatively resistant to MMS exposure and able to phosphorylate Rad53p. Unfortunately, we could not use mec1Δsml1Δsae2Δtel1Δ cells in tests for checkpoint function, as cells with this combination of mutations displayed a very early onset of senescence (see Fig. 6 for example). Thus, growth of mec1Δsml1Δsae2Δ cells expressing Tel1-11p was compared to mec1Δsml1Δsae2Δ cells expressing wild-type Tel1p in the presence of MMS (Fig. 6). At 26°C, mec1Δsml1Δsae2Δtel1-11 cells were no more sensitive to 0.01% MMS than mec1Δsml1Δsae2ΔTEL1 cells (Fig. 6, bottom left). At 37°C, mec1Δsml1Δsae2Δtel1-11 cells clearly were more sensitive to 0.01% MMS exposure than mec1Δsml1Δsae2ΔTEL1 cells (Fig. 6, bottom right).

At 37°C, the tel1-11 allele confers sensitivity to MMS exposure when combined with mec1Δsml1Δsae2Δ mutations. Top Growth tests in the absence of MMS by spot dilution were performed as described in Materials and methods. Cells used for these tests were grown for ∼30 generations before plating. About ten cells were plated on the most diluted spots. *Note the senescence of cells lacking all ATM-like kinases (mec1Δsml1Δsae2Δtel1Δ). Strains used are: 1 MCY7411-4B (mec1Δsml1Δsae2ΔTEL1); 2 MCY7412-18A (mec1Δsml1Δsae2ΔTEL1); 3 MCY7412-18B (mec1Δsml1Δsae2Δtel1-11); 4 MCY7422-7B (mec1Δsml1Δsae2Δtel1Δ); 5 MCY7422-7C (mec1Δsml1Δsae2Δtel1-11). Bottom Growth tests of strains in presence of MMS were performed as described in Materials and methods. About one cell was plated on the most diluted spots. Strains used are: 1 MCY7412-18A (mec1Δsml1Δsae2ΔTEL1); 2 MCY7412-18B (mec1Δsml1Δsae2Δtel1-11)
In a parallel assay, the cells were exposed to 0.01% MMS for a limited time (90 min, acute exposure) at 26 or 37°C and the extent of Rad53p phosphorylation was analyzed in total protein extracts from the cells exposed to these conditions. Phosphorylation was deduced from the appearance of slowly migrating forms of Rad53p using an anti-Rad53 antibody (Vialard et al. 1998). After acute exposure to MMS at 26°C, Rad53p was nearly completely phosphorylated in mec1Δsml1Δsae2ΔTEL1 cells and at least partially phosphorylated in mec1Δsml1Δsae2Δtel1-11 cells (Fig. 7). When these same cells were replated after the acute exposure on media without MMS at 26°C, a slight reduction in viability was observed for the mec1Δsml1Δsae2Δtel1-11 cells when compared to the in mec1Δsml1Δsae2ΔTEL1 cells (data not shown), which correlates well with the incomplete phosphorylation of Rad53p (Fig. 7). In contrast, at 37°C, Rad53p phosphorylation could only be detected in mec1Δsml1Δsae2ΔTEL1 cells, but not in mec1Δsml1Δsae2Δtel1-11 cells (Fig. 7) and the latter cells did not regrow on plates without MMS incubated at 37°C (data not shown). This correlation between analyses of re-growth and Rad53p-phosphorylation upon exposure to MMS demonstrates that in the genetic backgrounds used here, cells expressing Tel1-11p are severely compromised for the DNA-damage checkpoint at 37°C, while this function is quite proficient, when the assays were performed at 26°C.

Absence of detectable Rad53p phosphorylation in cells expressing Tel1-11p at 37°C. Phosphorylation of Rad53p in mec1Δsml1Δsae2ΔTEL1 or mec1Δsml1Δsae2Δtel1-11 cells exposed to 0.01% MMS for 90 min at 26 or 37°C. Assay conditions and detection of non-phosphorylated Rad53p (Rad53p) and phosphorylated Rad53p (Rad53p-P) was performed as described in Materials and methods. Strains used are: MCY7412-18A (mec1Δsml1Δsae2ΔTEL1); MCY7412-18B (mec1Δsml1Δsae2Δtel1-11)
Discussion
Tel1p has been implicated in both telomere maintenance and a DNA-damage checkpoint, sometimes referred to as TM-checkpoint (Greenwell et al. 1995; Usui et al. 2001). In addition, the MRX-complex has been associated with Tel1p for both of these functions (Tsukamoto et al. 2001; Usui et al. 2001). Although the first indication that these functions can be separated was provided by the discovery of an mre11-ts allele (Chamankhah et al. 2000), it remained unclear whether Tel1p contributes differentially to telomere maintenance and the DNA-damage checkpoint or whether its functions at telomeres are dependent on the same type and level of activity also required for the checkpoint. Here, we show that Tel1p contributes differentially to normal and basic telomere functions and DNA-damage checkpoints.
This conclusion is derived from the analyses of phenotypes conferred to cells by a new mutation in the TEL1 gene, called tel1-11. The mutation was isolated by virtue of its synthetic lethal interaction with a mec1-ts allele at 37°C. First, to assess the functions of tel1-11-encoded protein (Tel1-11p) in terms of maintenance of telomeric repeats, we analyzed two different phenotypes. The first relates to the ability of Tel1p to maintain a telomeric repeat tract of wild-type length, which we will refer to as the normal telomere function. The second is operationally defined as the activity which, in the absence of Mec1p, is required to maintain a stable telomeric repeat tract for more than 120 generations with the cells not undergoing a senescence crisis; we will call this activity the basic telomere function of Tel1p. Both of these functions are absent in cells completely lacking Tel1p (Figs. 4, 5). Intriguingly, Tel1-11p provides proficiency in both normal and basic telomere function when cells are grown at 26°C. However, in cells grown at 37°C, the mutation clearly affects normal telomere function, although not as severely as a tel1Δ mutation (Fig. 4). On the other hand and most significantly, in cells expressing Tel1-11p, the basic telomere function remains intact at all temperatures (Fig. 5). Second, we assayed the DNA-damage checkpoint functions conferred by Tel1-11p, as defined by phosphorylation of Rad53p and cell survival in a mec1Δsml1Δsae2Δ background upon exposure to MMS. Compared to cells expressing wild-type Tel1p, cells harboring Tel1-11p display a reduction of the level of Rad53p phosphorylation already at 26°C; and there is no phosphorylated Rad53p detectable, when the checkpoint functions are assayed in this setting at 37°C (Fig. 7). The sensitivities of these cells to DNA-damage induced by MMS parallel these findings quite well (Fig. 6 and data not shown). We conclude that DNA-damage checkpoint functions conferred by Tel1-11p are reduced at 26°C and virtually abolished at 37°C. These data are summarized in Table 2.
Hence and most strikingly, at 37°C, the basic telomere function of Tel1p is maintained by Tel1-11p even in the absence of detectable induction of a DNA-damage checkpoint. This differential requirement for Tel1p activity in terms of the basic telomere function and the checkpoint function can be rationalized in several ways. For example, these two functions could be carried out by different activities of Tel1p, or there are different targets of a common Tel1p activity, such as its kinase activity. In this latter scenario, Tel1-11p could be deficient in interactions required to activate the checkpoint, but still able to perform its activity on the targets required for maintaining telomeric repeats in the absence of Mec1p. Alternatively, the checkpoint and the basic telomere functions could have differential requirements for a common Tel1p activity on the same target. For example, even when the cells are grown at 37°C, Tel1-11p could supply a minimal kinase activity, just sufficient to phosphorylate a critical substrate to allow the basic telomere function. However, this minimal level of activity would not be sufficient to activate the DNA-damage pathway, at least in terms of detectable Rad53p phosphorylation or resistance to genotoxic stress. Although we do not know for certain whether the mutation in tel1-11 directly impairs Tel1p activity or causes decreased protein stability with normal levels of activity, at least HA3-tagged Tel1-11p protein levels expressed from the endogenous locus are very similar at 26 and 37°C, which is inconsistent with the mutation inducing a temperature-dependent protein instability (Supplemental Fig. 1).
The two hypotheses mentioned above are not mutually exclusive, since the mutation may actually affect different activities to different extents. The known genetic and physical interactions of Tel1p with other components of cellular checkpoint and repair mechanisms are consistent with either possibility, even though the MRX-complex has been placed in the same epistasis group as Tel1p with respect to its functions in the checkpoint as well as with respect to its functions at telomeres (Ritchie and Petes 2000; Usui et al. 2001), suggesting a common function. However, these genetic interactions may not necessarily reflect the same functional interactions. For example, recent data suggest that in vivo, the MRX-complex associates efficiently with DSBs in wild-type as in cells lacking Tel1p (Nakada et al. 2003a). The bound MRX-complex then interacts with Tel1p via a C-terminal domain of the Xrs2p, since the association of Tel1p with the breaks is dependent on this domain (Nakada et al. 2003a). On the other hand, telomeres isolated from asynchronously growing cells do not seem to be bound by the MRX-complex at an appreciable level, as long as Tel1p or Mec1p are present (Mieczkowski et al. 2003). Therefore, on telomeres, an activity that can be supplied by Mec1p or Tel1p appears to restrict and/or regulate the binding of the MRX-complex (Mieczkowski et al. 2003). As one possible interpretation of all the data, we speculate that the mutated Tel1-11p analyzed here may be severely impaired in its interactions at DSBs, but much less affected in its telomere-related activity. This would explain the drastic loss of the checkpoint activity while the basic telomere function remained intact in cells harboring this protein (Table 2).
Qualitatively, the degree of impairment of the normal telomere function conferred by Tel1-11p at 37°C lies between that required for the basic telomere functions and that required for checkpoint function, and this function is unaffected at 26°C, as discussed above (Table 2). These data reinforce the idea that the normal telomere function as well does not require a fully functional checkpoint (Morrow et al. 1995). However, given that in cells grown at 37°C, telomeric repeat tracts are not quite as long as in wild-type cells, it is difficult to predict whether this phenotype is caused by the impairment of the checkpoint activity or is the consequence of the mutation affecting another function of Tel1p. There is circumstantial evidence that Tel1p is involved in a Mec1-dependent, but MRX-independent G2/M-checkpoint signaling cascade (Giannattasio et al. 2002), and Tel1p has also been implicated in a chromosome capping function that prevents DSB to telomere fusions (Chan and Blackburn 2003). However, the latter issue is complicated by the fact that frequencies of direct telomere to telomere fusions are not increased in the absence of only Tel1p (Mieczkowski et al. 2003). It will be important to assess whether all of these functions depend on an active kinase function of Tel1p or whether some of them are dependent on other functions of Tel1p (see above).
Taken together, our data establish that Tel1p contributes differently to normal and basic telomere functions versus the DNA-damage checkpoint. This reinforces the idea that in wild-type cells, this yeast ATM-like kinase is involved in an important regulatory step during telomere replication and this function may be separable from the checkpoint function. Atm-lacking mammalian cells also display telomere-specific phenotypes, such as increased frequencies of chromosome end-fusions and accelerated loss of telomeric repeat DNA (Metcalfe et al. 1996; Pandita 2002). Given our results, it is possible that Atm-functions required at mammalian telomeres are different and separable from other cellular Atm functions. In fact, a recent phenotypic analysis of Terc −/− Atm −/− mice raised a similar possibility (Wong et al. 2003). Therefore, an in-depth analysis of separation-of-function alleles of ATM-like kinases in yeast could yield hints about the molecular defects in A-T patients.
References
Bosotti R, Isacchi A, Sonnhammer EL (2000) FAT: a novel domain in PIK-related kinases. Trends Biochem Sci 25:225–227
Brachmann CB et al (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14:115–132
Cha RS, Kleckner N (2002) ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297:602–606
Chakhparonian M, Wellinger RJ (2003) Telomere maintenance and DNA replication: how closely are these two connected? Trends Genet 19:439–446
Chamankhah M, Fontanie T, Xiao W (2000) The Saccharomyces cerevisiae mre11(ts) allele confers a separation of DNA repair and telomere maintenance functions. Genetics 155:569–576
Chan SW, Blackburn EH (2003) Telomerase and ATM/Tel1p protect telomeres from nonhomologous end joining. Mol Cell 11:1379–1387
Chan SW, Chang J, Prescott J, Blackburn EH (2001) Altering telomere structure allows telomerase to act in yeast lacking ATM kinases. Curr Biol 11:1240–1250
Clerici M, Paciotti V, Baldo V, Romano M, Lucchini G, Longhese MP (2001) Hyperactivation of the yeast DNA damage checkpoint by TEL1 and DDC2 overexpression. Embo J 20:6485–6498
Corda Y et al (1999) Interaction between Set1p and checkpoint protein Mec3p in DNA repair and telomere functions. Nat Genet 21:204–208
D’Amours D, Jackson SP (2001) The yeast Xrs2 complex functions in S phase checkpoint regulation. Genes Dev 15:2238–2249
D’Amours D, Jackson SP (2002) The mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol 3:317–327
Giannattasio M et al (2002) A dominant-negative MEC3 mutant uncovers new functions for the Rad17 complex and Tel1. Proc Natl Acad Sci USA 99:12997–13002
Gietz RD, Schiestl RH, Willems AR, Woods RA (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11:355–360
Goldstein AL, McCusker JH (1999) Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15:1541–1553
Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD (1995) TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell 82:823–829
Kranz JE, Holm C (1990) Cloning by function: an alternative approach for identifying yeast homologs of genes from other organisms. Proc Natl Acad Sci USA 87:6629–6633
Lisby M, Barlow JH, Burgess RC, Rothstein R (2004) Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118:699–713
Longhese MP, Paciotti V, Neecke H, Lucchini G (2000) Checkpoint proteins influence telomeric silencing and length maintenance in budding yeast. Genetics 155:1577–1591
Lundblad V, Blackburn EH (1993) An alternative pathway for yeast telomere maintenance rescues est1-senescence. Cell 73:347–360
Lundblad V, Szostak JW (1989) A mutant with a defect in telomere elongation leads to senescence in yeast. Cell 57:633–643
Lustig AJ, Petes TD (1986) Identification of yeast mutants with altered telomere structure. Proc Natl Acad Sci USA 83:1398–1402
McAinsh AD, Scott-Drew S, Murray JA, Jackson SP (1999) DNA damage triggers disruption of telomeric silencing and Mec1p-dependent relocation of Sir3p. Curr Biol 9:963–966
Metcalfe JA et al (1996) Accelerated telomere shortening in ataxia telangiectasia. Nat Genet 13:350–353
Mieczkowski PA, Mieczkowska JO, Dominska M, Petes TD (2003) Genetic regulation of telomere-telomere fusions in the yeast Saccharomyces cerevisae. Proc Natl Acad Sci USA 100:10854–10859
Morrow DM, Tagle DA, Shiloh Y, Collins FS, Hieter P (1995) TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell 82:831–840
Myung K, Chen C, Kolodner RD (2001) Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature 411:1073–1076
Nakada D, Matsumoto K, Sugimoto K (2003a) ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev 17:1957–1962
Nakada D, Shimomura T, Matsumoto K, Sugimoto K (2003b) The ATM-related Tel1 protein of Saccharomyces cerevisiae controls a checkpoint response following phleomycin treatment. Nucleic Acids Res 31:1715–1724
Nyberg KA, Michelson RJ, Putnam CW, Weinert TA (2002) Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet 36:617–656
Pandita TK (2002) ATM function and telomere stability. Oncogene 21:611–618
Ritchie KB, Petes TD (2000) The Mre11p/Rad50p/Xrs2p complex and the Tel1p function in a single pathway for telomere maintenance in yeast. Genetics 155:475–479
Ritchie KB, Mallory JC, Petes TD (1999) Interactions of TLC1 (which encodes the RNA subunit of telomerase), TEL1, and MEC1 in regulating telomere length in the yeast Saccharomyces cerevisiae. Mol Cell Biol 19:6065–6075
Rose MD, Winston F, Hieter P (1990) Methods in yeast genetics: a laboratory course manual. Cold Springs Harbor Laboratory Press, Cold Spring Harbor
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2 edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ (1996) Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271:357–360
Santocanale C, Diffley JF (1998) A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395:615–618
Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27
Takata H, Kanoh Y, Gunge N, Shirahige K, Matsuura A (2004) Reciprocal association of the budding yeast ATM-related proteins Tel1 and Mec1 with telomeres in vivo. Mol Cell 14:515–522
Takata H, Tanaka Y, Matsuura A (2005) Late S phase-specific recruitment of Mre11 complex triggers hierarchical assembly of telomere replication proteins in Saccharomyces cerevisiae. Mol Cell 17:573–583
Teng SC, Zakian VA (1999) Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol Cell Biol 19:8083–8093
Tsukamoto Y, Taggart AK, Zakian VA (2001) The role of the Mre11-Rad50-Xrs2 complex in telomerase- mediated lengthening of Saccharomyces cerevisiae telomeres. Curr Biol 11:1328–1335
Usui T, Ogawa H, Petrini JH (2001) A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell 7:1255–1266
Vialard JE, Gilbert CS, Green CM, Lowndes NF (1998) The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. Embo J 17:5679–5688
Weinert TA, Kiser GL, Hartwell LH (1994) Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev 8:652–665
Wellinger RJ, Wolf AJ, Zakian VA (1993) Origin activation and formation of single-strand TG1–3 tails occur sequentially in late S phase on a yeast linear plasmid. Mol Cell Biol 13:4057–4065
Wong KK et al (2003) Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature 421:643–648
Zakian VA, Scott JF (1982) Construction, replication, and chromatin structure of TRP1 RI circle, a multiple-copy synthetic plasmid derived from Saccharomyces cerevisiae chromosomal DNA. Mol Cell Biol 2:221–232
Zhao X, Muller EG, Rothstein R (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2:329–340
Zhu Y, Xiao W (2001) Two alternative cell cycle checkpoint pathways differentially control DNA damage-dependent induction of MAG1 and DDI1 expression in yeast. Mol Genet Genomics 266:436–444
Acknowledgements
We thank R. Rothstein, D. Durocher, T. Weinert, and M. -P. Longhese for generously providing yeast strains, plasmid constructs, or antibodies used in this study. This project was actually initiated by I. Dionne, whom we thank for providing unpublished data. The work was supported by the Canadian Cancer Society (NCIC research grant 013235). M. C. was supported by a postdoctoral fellowship by the Swiss National Science Fund and R. J. W. is a Chercheur National from the FRSQ.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Chakhparonian, M., Faucher, D. & Wellinger, R.J. A mutation in yeast Tel1p that causes differential effects on the DNA damage checkpoint and telomere maintenance. Curr Genet 48, 310–322 (2005). https://doi.org/10.1007/s00294-005-0020-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-005-0020-7