
Abstract
The Chlamydomonas reinhardtii chloroplast genome possesses thousands of small dispersed repeats (SDRs), which are of unknown function. Here, we used the petA gene as a model to investigate the role of SDRs in mRNA 3′ end formation. In wild-type cells, petA mRNA accumulated as a major 1.3-kb transcript, whose 3′ end was mapped to the distal end of a predicted stem-loop structure. To determine whether this stem-loop was required for petA mRNA stability, a series of deletions was constructed. These deletion strains accumulated a variety of petA mRNAs, for which approximate 3′ ends were deduced. These 3′ ends were found to flank stem-loop structures, many of which were formed partially or completely from inverted copies of SDRs. All strains accumulated wild-type levels of cytochrome f, demonstrating that alternative 3′ termini are compatible with efficient translation. The ability to form alternative mRNA termini using SDRs lends additional flexibility to the chloroplast gene expression apparatus and thus could confer an evolutionary advantage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chloroplast mRNA 5′- and 3′-untranslated regions (UTRs) carry important regulatory information. The 5′UTR associates with nucleus-encoded factors required for mRNA processing and stability and contains cis elements that function in translation initiation (for reviews, see Monde et al. 2000b; Zerges 2000). The 3′UTRs of most chloroplast mRNAs feature an inverted repeat (IR) that can fold into a stem-loop structure. These IRs do not terminate transcription, but instead serve as RNA-processing signals, with the mature 3′end flanking the IR (Stern and Gruissem 1987; Rott et al. 1996; Rott et al. 1998b). Disruption of the IR in vivo confers instability on transcripts (Stern et al. 1991; Lee et al. 1996; Monde et al. 2000a), because they become susceptible to 3′→5′ exonuclease activity (Drager et al. 1996). In addition, there is some evidence that 3′-processed mRNA is preferentially translated in Chlamydomonas chloroplasts (Rott et al. 1998a). Cleavage of the 3′ IR may also initiate mRNA decay. For example, depletion of the endoribonuclease CSP41, which preferentially recognizes RNA stem-loops (Bollenbach and Stern 2003), leads to slower degradation of some chloroplast transcripts in tobacco (Bollenbach et al. 2003). In summary, mRNA 3′ maturation is an important component of chloroplast gene expression and relies on the IR as a cis element.
The sequences and structures of 3′ IRs have been examined in great detail, using the spinach petD and Chlamydomonas atpB genes as models. Maturation of the petD pre-RNA became the first in vitro model for 3′ IR function, when its inefficient termination but rapid 3′→5′ exonucleolytic processing was revealed (Stern and Gruissem 1987). Using similar assays in Chlamydomonas, 3′ maturation of the atpB mRNA was found to be a two-step process, with endonucleolytic cleavage downstream of the IR followed by 3′→5′ exonuclease trimming (Stern and Kindle 1993). One of the enzymes involved is likely to be polynucleotide phosphorylase, since Arabidopsis plants partially depleted for PNPase accumulate certain 3′-unprocessed chloroplast transcripts (Walter et al. 2002). Other proteins are undoubtedly involved as well, for example the Chlamydomonas nuclear mutant crp3 has multiple deficiencies in chloroplast RNA 3′ maturation (Levy et al. 1997). Furthermore, several 3′ IRs have been shown to bind specific proteins in vitro (Chen et al. 1995; Hayes et al. 1996; Memon et al. 1996), although only in one case has a function been tentatively assigned (Schuster and Gruissem 1991).
In spite of this importance, the IR primary sequence has little effect on gene expression. For example, the 3′ IR of the Chlamydomonas atpB or uidA reporter mRNAs can be replaced in vivo by those of other plastid genes, such as spinach petD, rbcL, petA, and Escherichia coli thrA (Blowers et al. 1993; Stern and Kindle 1993; Rott et al. 1998b), or by a polyguanosine sequence, which can form a tertiary structure (Drager et al. 1996). There is also little effect on β-glucuronidase (GUS) accumulation in tobacco chloroplast transformants when the 3′ IR is exchanged (Staub and Maliga 1994), although the lack of an IR strongly reduces GUS expression (Monde et al. 2000a). The importance of structure, but not sequence, is typical of prokaryotic 3′UTRs and would appear to simplify the coordination of gene regulation.
The recently completed sequence of the Chlamydomonas chloroplast genome (Maul et al. 2002) offers the opportunity to examine 3′ IR function in a broader sense. Perhaps the most remarkable feature of this genome is the profusion of small dispersed repeats (SDRs), of which more than 20,000 populate most of the intergenic regions. Whether the SDRs have any function in genome replication, stability, or gene expression is unknown, apart from a report associating an SDR-rich region upstream of petA with light-regulated transcriptional regulation (Thompson and Mosig 1987). The petA–petD intergenic region, for example, is rich in SDRs, yet contains the petA mRNA 3′ end (Matsumoto et al. 1991) and a 112-amino-acid open reading frame (ORF112) of uncertain functionality (Büschlen et al. 1991). Given the unknown function(s) of SDRs and the fact that only in the case of Chlamydomonas atpB has the 3′ IR of a chloroplast gene been manipulated in its native context (Stern et al. 1991), the petA gene is an attractive target. Here, we report the results from a variety of deletions in the petA–petD intergenic region, with the major conclusion being that some, but not all SDRs are able to form stem-loop structures and thus create alternative 3′ ends for chloroplast transcripts.
Materials and methods
Strains and culture conditions
The wild-type strain used in this study was P17 (Stern et al. 1991), which is derived from CC373, an atpB deletion mutant (Shepherd et al. 1979). The strain ΔpetA, in which the petA gene has been replaced by an aadA selectable marker cassette (Kuras and Wollman 1994), was used as recipient for chloroplast transformation to create the ΔF mutant series. Chlamydomonas cells were grown in Tris/acetate/phosphate (TAP) medium (Harris 1989) at 23 °C with 50 μE m−2 s−1 fluorescent light under a 18/6 h light/dark cycle. Chloroplast transformation was carried out as described by Kuras and Wollman (1994), with selection on minimal medium requiring photoautotrophic growth. The transformants were colony-purified and the expected deletions were confirmed by PCR and DNA filter hybridizations.
Plasmids
The plasmid pWF (Kuras and Wollman 1994), which contains the whole petA gene and extends into the petD-coding region, was linearized with EcoRV, which cleaves near the mature petA mRNA 3′ end. A time-series of deletions was created by treatment with exonuclease III as described by Maniatis et al. (1989). For most clones, the ends were blunted with T4 DNA polymerase and rejoined by ligation. For the clone Δ44, the 3′ deletion product was gel-purified and religated to the proximal part of EcoRV-linearized pWF, generating a unidirectional deletion. Plasmids were purified from various exonuclease III deletion time-points and analyzed by PCR to ensure that they contained sufficient petA-coding sequences to confer photoautotrophic growth, assuming that the mRNA accumulated. Subsequently, the exact length of deletion was determined by dideoxy nucleotide-sequencing. In most cases, the extents of 5′ and 3′ deletion were unequal and sometimes remarkably so, yielding a variety of intergenic remnants.
Protein and RNA analysis
Total protein and RNA were isolated from strains grown in TAP liquid medium as described by Drager et al. (1996, 1998). Immunoblot analysis of cytochrome f and the ATPase β-subunit with enhanced chemiluminescence was as described by Higgs et al. (1999). Immunoblotting signals were scanned and quantified using ImageQuant software and a Storm scanner (Molecular Dynamics, Sunnyvale, Calif.). Total RNA was separated in 1.2% agarose/3% formaldehyde gels, blotted onto Nylon membranes, and hybridized with 32P-labeled probes as described by Drager et al. (1998) and Higgs et al. (1999). Radioactive bands were visualized with a Storm Scanner.
Reverse transcriptase–polymerase chain reaction
Total RNA (2–5 µg) was first treated with 2–5 units RQI DNase I (Promega) for 5 min at 37 °C and then purified by phenol/chloroform extraction and ethanol precipitation. RNA (0.5 μg) was ligated to 250 ng oligoribonucleotide tail, using 10 units T4 RNA ligase at 15 °C for 12 h (see Table 1 for primer sequences). The RNA tailed-transcript pool was reverse-transcribed, using an adapter primer complementary to the RNA tail, and cDNAs containing petA 3′ ends were amplified by PCR using petA-specific primers a and b and the adapter (Fig. 4, Table 1). The PCR products were inserted into the vector pGEM-T Easy (Promega) and analyzed by DNA sequencing.
Computer analysis of repeated elements
In this study, we used four Web-based sequence analysis tools: Mfold, Palindrome, PipMaker, and RepeatFinder. Mfold (Zuker 2003) was used to fold sequences of the defined or derived petA 3′ ends with default settings at a temperature 37 °C. Mfold often generates alternative folding patterns; and the ΔG values of individual stem-loops in Fig. 5A were determined by analyzing only the stem-loop, rather than a longer sequence. Palindrome, located at http://bioweb.pasteur.fr/seqanal/interfaces/palindrome.html, allows users to look for IRs in candidate sequences and to compute alignments and mismatches. The output is a list of aligned IR sequences with their starting and ending positions, but no loop sequences. Here, we analyzed the 2,530-bp petA–petD intergenic region plus flanking genic sequences and compared this output to that of Mfold. This was used to develop the IR map shown in Fig. 5A.
PipMaker (Schwartz et al. 2000) allows users to compare large DNA sequences and identify regions of high sequence similarity, using Blast. PipMaker tools can be found at http://bio.cse.psu.edu/pipmaker/. Here, the petA–petD intergenic sequence was plotted against itself, or against the complete Chlamydomonas chloroplast genome (GenBank accession number BK000554) from which the 2,530-bp region had been specifically deleted. The output percentage identity plots for these two alignments are presented in Fig. 5B. Another tool, RepeatFinder, located at http://cbsu.tc.cornell.edu, allows user-definable parameters and generates a database of repeated elements. Using as input the Chlamydomonas chloroplast genome and default parameters, the output file containing the extracted SDRs (clustered in groups) was used to search homologous sequences to the 3′ end IR or stem sequences. The results (matched SDR families with the number of their members) are presented in Table 2.
Results
Deletion mutants in the petA–petD intergenic region
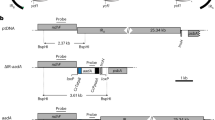
The petA gene is located approximately 1.7 kb downstream of the disabled Wendy transposon, near the defined origin of the 203-kb chloroplast genome (Maul et al. 2002). As shown in Fig. 1A, ORF112 lies 328 bp downstream of petA and the petD transcription unit is separated from ORF112 by an additional 1.5 kb. Using a plasmid covering both petA and the 5′ end of the petD region, a series of deletion constructs was made, following linearization at the EcoRV site just upstream of ORF112 (Fig. 1A; see Materials and methods). Because we wished to study the role of RNA elements in petA expression, deletion clones were screened for those which, assuming sufficient RNA accumulation, would encode the functional cytochrome f. Thus, the furthest upstream 5′ deletion endpoint used was in Δ30 (Fig. 1B), based on the known C-terminal functional domains of cytochrome f (Choquet et al. 2003). Δ30, along with other constructs selected to represent a range of deletion endpoints, was used for chloroplast transformation with strain ΔpetA as a recipient. In ΔpetA, the petA gene had been replaced with the aadA selectable marker cassette, conferring an acetate-requiring phenotype (Kuras and Wollman 1994). Following transformation, colonies were selected on minimal medium, where cells had to grow photoautotrophically. Each of the strains for which a deletion is represented in Fig. 1B generated multiple colonies on this medium, suggesting that some accumulation of the cytochrome b 6 /f complex was occurring. The transformants were subsequently verified by PCR and DNA filter hybridization analysis (data not shown).

Diagram of the petA–petD region and deletion mutants in the chloroplast genome of Chlamydomonas reinhardtii. A A HincII/HindIII fragment covers a region of 3.8 kb including the petA gene and the 5′UTR of petD. The black and shaded boxes indicate the petA and ORF112 open reading frames, respectively, and the arrows their directions of transcription. Bent arrows indicate the locations of transcript 5′ termini. The 3.8-kb fragment was subcloned into pHcRV-1.6 and pCD-RV-H2.2, shown below. Indicated restriction sites are: H HindIII, Hc HincII, RV EcoRV, Afl AfllI, Avr AvrII. B A segment expanded from panel A is shown. The horizontal lines represent the deletions carried in the chloroplast genomes of different ΔF strains (see Materials and methods). The numbers before and after the lines are the nucleotide endpoints of each deletion, with position 1 being the final A of the petA translation termination codon. WT Wild type
The 11 mutants for which transformants were generated are shown in Fig. 1B, along with their precise deletion endpoints. Three mutants (Δ6, Δ16, Δ44) had long deletions, whereas the other eight mutants had relatively short deletions surrounding the EcoRV site. All deletions except Δ69 affected ORF112, demonstrating that it is not essential for photosynthesis. However, many of the deletions removed the normal 3′ maturation site for the major petA transcript (see: Altered petA mRNA profiles in the mutant strains).
The deletion mutants accumulate near-wild-type levels of cytochrome f
Since transformants were selected on minimal medium, they were evidently photoautotrophic. Although the cytochrome b 6 /f complex is essential for photosynthesis, cells are conditionally photoautotrophic with as little as 10% of the accumulating complex (Chen et al. 1993, 1997). This mirrors similar work on the atpB gene, which showed that deletion of the atpB 3′ end IR destabilized atpB mRNA and led to approximately 10% of wild-type ATPase β-subunit protein accumulation (Stern et al. 1991). As for the cytochrome b 6 /f complex, the atpB deletion was conditionally acetate-requiring (Levy et al. 1997).
In the case of the mutants described here, however, there was no evidence for temperature sensitivity (data not shown). We therefore examined accumulation of cytochrome f using immunoblot analysis, with the ATPase β-subunit as a loading control; and a representative blot is shown in Fig. 2A. Using quantification of proteins from several independent preparations, we concluded that cytochrome f accumulated at normal levels in all deletion strains analyzed (Fig. 2B). This result led us to ask what form(s) of petA mRNA accumulated in the mutants which could support production of normal cytochrome f levels.

Immunoblot analysis of ΔF strains. A Wild-type and mutant strains were grown in Tris/acetate/phosphate medium at 23 °C under 50 µE m−2 s−1 light and a 18/6 h light/dark cycle. Total protein (10 µg) from each strain was fractionated by SDS-PAGE, transferred to a polyvinylidene difluoride membrane, and challenged with antibodies directed against the indicated proteins. Numbers across the top indicate the ΔF deletion strain used. B Two strains for each deletion construct were analyzed in multiple experiments and immunoblotting signals were quantified. The amount of cytochrome f was normalized to the ATPase β-subunit and displayed as a fraction of the cytochrome f in the wild-type cells. T-tests showed that there were no significant differences in the amount of cytochrome f between wild-type cells and the ΔF mutants (n=4, P=0.05)
Altered petA mRNA profiles in the mutant strains
Total RNA was isolated from duplicate transformants of each deletion construct, and filter hybridization was performed with probes corresponding to the petA- and petD-coding regions, with atpB used as a loading control. Figure 3A shows that discrete petA transcripts accumulated in each strain, but that the profile was altered in each of the mutants, while neither the petD profile nor its accumulation relative to the atpB were altered. The wild-type has a major petA transcript of 1.3 kb, with minor bands at 1.5–1.6 kb and 3.4 kb. In comparison, six mutant strains (Δ1, Δ2, Δ4, Δ57, Δ62, Δ69), each of which contained short deletions adjacent to the EcoRV site or the ORF112 initiation codon, accumulated species of 1.3 kb, 1.6–1.7 kb, and 3.0–3.4 kb. The most obvious difference in comparison with the wild-type was that the 1.3-kb transcript was decreased in the mutants, while the band at 1.6–1.7 kb became dominant. This suggests that a region impacted in each of these deletions is required for efficient 3′ end formation of the 1.3-kb species.

RNA filter hybridization analysis of petA, petD, and atpB mRNAs. A Cells were grown as described in the legend to Fig. 2. Total RNAs (5 µg lane−1) were fractionated in a 1.2% agarose gels and probed with the genes indicated at left. Transcript sizes were estimated using RNA markers from Invitrogen. B Diagram showing the inverted repeat (IR) structure ending 77 nt downstream of the translation termination codon in the wild-type petA sequence and its relationship to the deletion endpoints in strains Δ6, Δ7, and Δ30. Position 2178 is the petD mRNA 5′ end
The other five mutant strains (Δ6, Δ7, Δ16, Δ30, Δ44) had still more dramatic changes in their petA transcript profiles. Consistent with its deletion spanning most of the petA–petD intergenic region, Δ6 had a single, short transcript of 1.4 kb, while Δ16 and Δ44 still accumulated the wild-type 1.3-kb transcripts, but lacked the 3.4-kb species. Δ16 also reproducibly accumulated heterodisperse RNAs migrating between 1.5–1.8 kb and 2.5–2.8 kb, while Δ44 had a major band at 1.6 kb. Although both Δ7 and Δ30 had relatively short deletions, they lacked the 1.3-kb wild-type transcript, suggesting that 3′ end formation signals had been deleted. Instead, Δ7 had two major transcripts of 1.4 kb and 1.7 kb and disperse transcripts near 2.8 kb; and Δ30 accumulated transcripts of 1.6 kb and 3.0 kb. Taken together, the various mutants exhibited both qualitative and quantitative changes in petA-hybridizing RNAs.
To interpret cases where the major wild-type 1.3 kb transcript had been lost, we predicted RNA-folding using Mfold 3.1 (Zuker 2003). This revealed that a perfect stem-loop can be formed by sequences between 41 nt and 77 nt downstream of the petA translation termination codon, as shown in Fig. 3B. The deletions in Δ6, Δ7, and Δ30 had destroyed or eliminated this stem-loop and lost the 1.3-kb wild-type petA mRNA, which suggests that this stem-loop is its 3′ maturation site. This prediction is in accordance with the size calculated by Matsumoto et al. (1991) and RT-PCR mapping (see next section). In the cases where the 1.3-kb RNA was reduced in relative abundance without affecting the stem-loop, we suggest that efficiency of use rather than RNA stability was the cause.
RT-PCR mapping of petA mRNA 3′ ends
To amplify petA 3′UTR sequences, we performed PCR with petA-specific primers a and b in combination with a DNA adapter (Fig. 4; Table 1) after RNA tail ligation and reverse transcription. This 3′RACE method amplified a fragment (~250 bp) in the wild-type sample at annealing temperatures of both 55 °C and 58 °C, using primer a. A similar band could be amplified from other mutants which accumulated the 1.3-kb transcript. The PCR fragment was cloned and sequenced; and multiple clones from several strains showed that the RNA tail had been ligated 77 nt downstream of the petA translation termination codon, at the distal end of the predicted stem-loop structure (Fig. 3B). Additional 3′RACE clones were generated using petA primer b, which when sequenced identified a number of minor 3′ ends, including some which were consistent with the minor bands at 1.5–1.7 kb (Fig. 3A). However, by far the most frequent 3′UTR site identified was at the +77 position.

RNA filter hybridization analysis to map approximate petA mRNA 3′ ends. Total RNA and filter blots were prepared as described in the legend to Fig. 3. The top of the figure shows a map of the petA–petD region, with the locations of probes used in the blots below (dark lines) or a probe that did not give useful results (gray line; see text). Restriction sites are as in the legend to Fig. 1. Arrowheads above represent the primers listed in Table 1, which were used to generate the probes, or in RT-PCR. The bottom four panels show hybridizations with the probes indicated below each one. Strains are labeled across the top and RNA sizes (in kilobases) are shown at the right. Species marked by arrowheads in blot i-j are discussed in the text. These have the same 3′ end as the species at 3.0–3.4 kb in other strains. A rRNA hybridization artifact is marked with an asterisk
In an effort to confirm a possible association between SDRs and 3′ end formation, it was desirable to obtain as much 3′ end fine-mapping data as possible. At the same time, the presence of SDRs offered technical challenges, with both 3′RACE and S1 nuclease protection. Despite numerous efforts, the high background in S1 protection made it impossible to map petA 3′ termini using this method (data not shown).
RNA-walking to approximate 3′ ends of larger petA transcripts
The 3′ termini of RNAs accumulating in the deletion mutants could be roughly predicted, knowing the size of deletion, and assuming that the 5′ ends were the same as in wild-type cells. To correlate such predictions experimentally, we performed filter hybridizations with several probes spanning the intergenic region, as shown at the top of Fig. 4. The Avr-H probe proved unusable, perhaps due to the presence of repeat elements. Probe e-f contains ORF112 and only hybridized with transcripts of ≥1.6 kb, placing the 3′ ends of the shorter species between the petA-coding region and the 5′ end of this probe. Δ6 lacks the e-f region and displayed only background hybridization, probably due to repeat elements; and some of these indistinct bands also appeared in other lanes. Several strains had transcripts of 1.6–1.7 kb which hybridized strongly. Based on their lengths and size of deletions, these species terminate downstream of ORF112. The small migration differences, for example between Δ1 and Δ2, are consistent with differences in the extent of their deletions. As expected, the longest transcripts (>3 kb) hybridized with probe e-f.
Probe g-h, covering the gap between e-f and probe Afl-Avr, hybridized with transcripts of 1.6–1.7 kb, except in Δ6, Δ16, and Δ44, where the g-h region was deleted. In contrast, probe Afl-Avr failed to hybridize with them, except in the case of Δ7. Taken together, this serves as further evidence that the 3′ ends of the transcripts at 1.6–1.7 kb lie downstream of ORF112 and upstream of the AflII site defining the 5′ end of probe Afl-Avr. As expected, the longest transcripts (>3 kb) hybridized with probe g-h, except in strains where this region was deleted.
As noted above, Δ7 was exceptional in that its 1.7-kb petA transcript hybridized with both probes g-h and Afl-Avr. As shown in Fig. 1B, the 3′ end of the Δ7 deletion lies further downstream than that in all other deletion strains, except Δ6/Δ16/Δ44. Given the probe and the deletion, the Δ7 1.7-kb RNA would be expected to hybridize only weakly with the e-f probe, as is the case. The simplest conclusion from the hybridization data is that Δ7 lacks the 3′ end formation signals that generate the termini of the other transcripts at 1.6–1.7 kb seen, for example, with the g-h probe. Therefore, the 3′ end of the Δ7 transcript lies downstream of the AflII site. Its apparent abundance suggests that this is an efficient 3′ end formation signal and/or strong stability determinant.
To determine the extents of the transcripts at 3.0–3.4 kb, we first used probe Avr-H, which proved to hybridize with many bands (data not shown). Therefore, we generated a shorter probe, i-j, which resulted in a relative simple pattern of hybridization (Fig. 4, lower right panel). The petA transcripts identified were the species at 3.0–3.4 kb (2.8 kb in Δ7) in all strains, except Δ6, Δ16 and Δ44, suggesting that these longer RNAs terminated near the petD transcription initiation site. In Δ6, Δ16, and Δ44, which have long deletions, the hybridizing species were 1.4 kb, 2.5 kb, and 1.6 kb, respectively (marked by arrowheads). This is consistent with a 3′ end similar to the other strains. In addition, probe i-j hybridized with a 1.8-kb RNA in all lanes. We believe this to be rRNA, since several segments in the probe sequence share similarity with rRNA sequences (data not shown).
The mapped or deduced petA mRNA 3′ ends are close to SDR-containing stem-loops
By RNA filter hybridizations, RT-PCR, and S1 nuclease protection, we analyzed and mapped several possible 3′ ends of petA transcripts. The 3′ end of the major 1.3-kb transcript was defined by all three approaches, whereas others had to be derived, as shown in Table 2. To do so, we first measured as accurately as possible transcript sizes on filter blots, using molecular mass markers. We then assumed that the mRNA 5′ ends did not deviate from the wild type. For example, three closely clustered 5′ termini were mapped by Matsumoto et al. (1991). Knowing the exact sizes of deletions, we could then calculate the location of the 3′ ends, relative to the +1 position defined in Fig. 1C. We estimate our accuracy to be ±20 nt.
To determine whether these mapped or deduced 3′ ends coincided with stem-loop-forming IR sequences, we analyzed the petA–petD intergenic region, using the programs Palindrome and Mfold 3.1 (see Materials and methods). This revealed a large number of IRs that could serve as petA mRNA 3′ ends, based on the transcript-sizing discussed above. Those at or near locations predicted for 3′ ends are diagrammed in Fig. 5A with respect to the wild-type sequence, along with the strains in which those ends are found (major ends for each strain are underlined). The sequences are also given in Table 3, to show their relationships to SDRs (see below). The IR defining the 1.3-kb 3′ end is fairly AU-rich, with a predicted ΔG value of –10.3 kcal mol−1, whereas the IR defining the ≥3-kb RNA 3′ ends harbors a long, perfect repeat, with a ΔG of −58 kcal mol−1. This IR is located 200 bp upstream of the petD mRNA 5′ end and somewhat closer to the petD promoter, since the mature 5′ end is derived from the processing of a longer precursor (Sakamoto et al. 1994). Similarly, IR sequences are found at or near each of the intervening 3′ ends (RNAs) of 1.5–1.8 kb. How precisely the mRNA 3′ ends correspond to the IR 3′ ends cannot be determined.

Percentage identity plots of small dispersed repeat (SDR)-containing stem-loops associated with petA mRNA 3′ ends. A Stem-loops mapped at petA 3′ ends, which contain the IR sequences listed in Table 3, are shown above the petA–petD intergenic region. For each stem-loop, its ending nucleotide position is labeled; and strains which appear to utilize each stem-loop are indicated at the side. The stem-loop associated with the most abundant mRNA, based on hybridization with a petA coding region probe, is underlined for each strain and highlighted in Table 2. B SDRs in a 2.53-kb sequence covering the region between the petA translation termination codon and the petD translation initiation codon are shown, following sequence analysis using PipMaker. In the top panel, this 2.53-kb sequence was analyzed for IRs, which are arranged horizontally according to their position in the intergenic region and vertically according to percent identity with the reference sequence, beneath which each aligned repeat is placed. The length of the line represents the length of the identity. The percent identity is from 100% to 50%, as shown at the right. In the middle panel, the intergenic region was queried against the remainder of the chloroplast genome. The increased density of repeats reflects the fact that repeats present in the intergenic region are also present in many other locations. These locations are depicted in the bottom panel, which is a dot-plot of the intergenic region against the full chloroplast genome
The IRs shown in Fig. 5A are necessarily derived from repeated sequences (i.e. the two stems), but we wished to determine whether those repeats belong to the genome-wide SDRs. A rapid scan of such repeat elements can be derived using the web-based PipMaker program (see Materials and methods). As we showed previously on a whole-genome level (Maul et al. 2002) and show in more detail in Fig. 5B, the petA–petD intergenic region harbors >1,000 SDRs, which contrasts with their virtual absence within the petA and petD genes. Figure 5B shows three such representations. In the top panel, repeats were searched using the intergenic region alone; and this shows the prevalence of repeats, excluding the remainder of the genome. In the middle panel, the intergenic region was searched against the rest of the genome. Since each line represents a copy of some intergenic sequence present elsewhere, it is obvious that petA–petD intergenic sequences are repeated in multiple locations outside this immediate region. A general idea of these locations can be gained from the bottom panel of Fig. 5B. In this panel, the linearized chloroplast genome is represented from left to right. The dots below it represent the locations of repeated petA–petD intergenic sequences, of which there are >40. Whether these widely dispersed copies might form other mRNA 3′ termini is under investigation.
To attempt to match the IR sequences in Fig. 5A and Table 3 with SDRs, we used the RepeatFinder program to generate a SDR database (see Materials and methods). Interestingly, we found that all the IR sequences mapped to petA RNA 3′ ends include, or consist entirely of, SDRs. Table 2 shows the SDR family numbers and their sizes, which range from 10 to 83 members; and Table 3 shows the precise relationship of SDRs to the IR sequences themselves. Taken together, these data show that some SDRs can form IR sequences that likely serve as mRNA 3′ end maturation sites.
Discussion
Consequences of petA 3′UTR deletions
We created a deletion series downstream of the petA translation termination codon, where sequence analysis shows more than ten IRs within the following 300 bp. Nevertheless, apart from the major 3′ end IR, petA transcripts mature other IRs rarely, or weakly if at all. This implies a selectivity in the processing mechanism, which must recognize both sequence and secondary structure. One possibility is that petA is processed by a two-step endonuclease–exonuclease mechanism, as is atpB (Stern and Kindle 1993), and one particular IR serves as a maturation site because the endonuclease recognizes a sequence between this IR and the next downstream. Deletions into the petA 3′UTR, including those eliminating ORF112 and nearly all of the petA–petD intergenic region did not affect photosynthetic growth, although in some cases they radically changed the profiles of petA mRNAs. The results were surprisingly different from similar studies with the atpB gene, where analogous deletions caused weak or non-photosynthetic phenotypes (Stern et al. 1991). The reason for this was revealed by RNA analysis, which demonstrated that petA transcripts can mature at locations further downstream; i.e. different petA 3′UTR IRs can be interchangeable. This is consistent with results obtained in both Chlamydomonas and tobacco chloroplast transformants, where the 3′ IR of one gene can generally be replaced by another, preferably in the sense orientation (Blowers et al. 1993; Staub and Maliga 1994; Rott et al. 1998b; Eibl et al. 1999). However, the wide variation in the accumulation of different species suggests that not all petA 3′ IRs are functionally equivalent. Possible reasons are the variable thermodynamic stabilities of the stem-loops (Table 3), or preferential endonuclease cleavage, as discussed above.
Sequence and structural complexities of petA mRNA 3′ end-mapping
We used three different approaches to map petA 3′ ends. The major 3′ end (of the 1.3-kb species) was mapped precisely and found to be 77 nt downstream of the translation termination codon, coinciding with an IR. The importance of the IR was further confirmed by analysis of petA transcripts in deletion strains Δ6, Δ7, and Δ30 (see Figs. 1, 3). Two-thirds of the IR are missing in Δ6 and Δ7, whereas the Δ30 deletion completely eliminated the IR. These three strains lacked the 1.3-kb transcript, whereas deletion strains which maintained this IR accumulated it. Our mapped location of the major petA 3′ end is 80 nt upstream of a site reported by Matsumoto et al. (1991). We ascribe this discrepancy to the very large probe used for S1 nuclease protection, which yielded a difficult-to-size protected fragment (~1,025 nt) in their Fig. 5. The same 3′ end was apparently also mapped 213 nt downstream of the translation termination codon, using a RNase protection protocol (Rott et al. 1996). We believe these data can be reinterpreted as either mapping one of the minor transcripts at 1.5–1.7 kb (Fig. 3A), or it may be an artifact of the method used.
We encountered continual difficulties in mapping additional petA 3′ ends in the SDR-rich intergenic region. S1 protection was likely confounded by the tendency of the probe to self-anneal, giving a background of numerous protected bands, despite efforts to optimize the hybridization temperature. RT-PCR was complicated by the paucity of unique sequences that could be used for primer design and the low efficiency of T4 RNA ligase-mediated attachment of adapter primers to highly structured transcripts. This forced us to rely on precise transcript-sizing to estimate 3′ ends, along with “Northern walking” and knowledge of deletion endpoints. While this reduced the accuracy of 3′ end-mapping, it is typical of what can be expected in such repeat-rich situations.
Site preference for 3′ end formation
Transcript stoichiometries varied widely, suggesting that 3′ end selection is not simply determined by the presence of a given IR. For example, deletions in Δ1, Δ2, Δ4, Δ57, Δ62, and Δ69 significantly enhanced accumulation of the transcripts at 1.6–1.7 kb, although the original IR defining the 1.3-kb mRNA 3′ end remained intact (Figs. 1, 3A). The 3′ ends of these longer transcripts were mapped at 660–850 nt downstream of the petA translation termination codon, in a region rich in IRs (Fig. 5A). The preferential use of these sites in the deletion mutants mentioned above suggests a 3′ end selection mechanism that disfavors the normal wild-type 3′ end. One possibility is that, following transcription and presumably inefficient termination (as has been repeatedly shown for chloroplast IRs; Stern and Gruissem 1987; Rott et al. 1996), an initial endonucleolytic cleavage step takes place preferentially downstream of the petA region at 660–850 nt, rather than between this region and the favored IR in wild-type cells. Another alternative is that the 3′ ends of the transcripts at 1.6–1.7 kb are formed directly by transcription termination, and inefficient processing to generate the 1.3 kb species allows this to occur at an increased frequency.
A third region where petA mRNA ends formed in all strains is represented by the 3.4-kb transcript in wild-type cells, with correspondingly shorter lengths in deletion strains. We estimated the maturation site to be approximately 200 nt upstream of the petD mRNA 5′ end. This raises the interesting possibility that the petA 3′ end maturation at this site may be related to petD 5′ end maturation. We showed that the petD 5′ end is formed through the processing of a primary transcript that initiates within 100 nt upstream, but can also be formed by the processing of a petA–petD dicistronic transcript (Sturm et al. 1994). In the latter scenario, exonucleolytic resection of an initial processing product could yield both mature petD mRNA and the 3.4-kb petA transcript. This mechanism is likely to be responsible, at least in some cases, for generating shorter transcripts from polycistronic primary transcripts of higher plant chloroplast gene clusters. Arguing against this, however, is the case of the Arabidopsis mutant hcf107, which has a defect in processing the 5′ end of psbH mRNA, but accumulates normally upstream transcripts whose 3′ ends might be concomitantly formed (Felder et al. 2001). Thus, whether endonucleolytic cleavages that form 5′ and 3′ ends are interdependent, or independent, remains to be established.
3′ IRs of petA mRNAs are members of SDRs
The petA–petD intergenic region, like most in the Chlamydomonas chloroplast, is heavily populated with repeat sequences (Maul et al. 2002); and such repeat-infested regions in chloroplasts have been hypothesized to be mutational hotspots and promote genome rearrangements (Palmer et al. 1985; Palmer 1991; Boynton et al. 1992; Newman et al. 1992; Morton and Clegg 1993; Cosner et al. 1997). The petA–petD intergenic region is saturated with SDRs, as shown in Fig. 5B, in contrast to the adjacent protein-coding regions. Because all the petA 3′ ends mapped in this study appear to be associated with inverted SDRs, it could be argued that selection is acting to maintain these SDR copies as a mechanism to provide alternative functional 3′ maturation sites. Equally, one could contend that utilization of SDRs is a consequence of their presence and confers no selective advantage. Because deletion of the Chlamydomonas atpB or psaB 3′ IRs causes a photosynthetically impaired phenotype (Stern et al. 1991; Lee et al. 1996), redundancy could clearly be advantageous in some circumstances. In a more general sense, nucleus-encoded mRNAs are often found with multiple polyadenylation sites (Legendre and Gautheret 2003), which can have substantial consequences for gene regulation (Cui et al. 2003). Although we previously reported that 3′ end-processing facilitates translation in Chlamydomonas chloroplasts (Rott et al. 1998a), we have not yet determined whether the petA transcripts described here are differentially translated, or have different stabilities.
It is also unclear how many Chlamydomonas chloroplast transcripts have 3′ termini coinciding with SDRs. Only a few 3′ ends have been definitively mapped, in particular those of atpB, petD, psaB, and rbcL (Stern et al. 1991; Blowers et al. 1993; Sakamoto et al. 1993). Each of these 3′ ends corresponds to an IR; and each of these IRs contains sequences also found in SDRs, both in small (10–20 members) and large families (data not shown). At the least, this points to petA as not being a unique case and suggests that further association of SDRs with RNA 3′ ends (and possibly other gene expression functions) is likely to be uncovered.
References
Blowers AD, Klein U, Ellmore GS, Bogorad L (1993) Functional in vivo analyses of the 3′ flanking sequences of the Chlamydomonas chloroplast rbcL and psaB genes. Mol Gen Genet 238:339–349
Bollenbach TJ, Stern DB (2003) Secondary structures common to chloroplast mRNA 3′-untranslated regions direct cleavage by CSP41, an endoribonuclease belonging to the short chain dehydrogenase/reductase superfamily. J Biol Chem 278:25832–25838
Bollenbach T, Tatman D, Stern D (2003) CSP41, a mutltifunctional RNA-binding protein, initiates mRNA turnover in tobacco chloroplasts. Plant J 36:842–852
Boynton JE, Gillham NW, Newman SM, Harris EH (1992) Organelle genetics and transformation in Chlamydomonas. In: Hohn T (ed) Cell organelles. Springer, Berlin Heidelberg New York, pp 364–389
Büschlen S, Choquet Y, Kuras R, Wollman F-A (1991) Nucleotide sequences of the continuous and separated petA, petB, and petD chloroplast genes in Chlamydomonas reinhardtii. FEBS Lett 284:257–262
Chen Q, Adams CC, Usack L, Yang J, Monde R, Stern DB (1995) An AU-rich element in the 3′ untranslated region of the spinach chloroplast petD gene participates in sequence-specific RNA–protein complex formation. Mol Cell Biol 15:2010–2018
Chen X, Kindle K, Stern D (1993) Initiation codon mutations in the Chlamydomonas chloroplast petD gene result in temperature-sensitive photosynthetic growth. EMBO J 12:3627–3635
Chen X, Simpson CL, Kindle KL, Stern DB (1997) A dominant mutation in the Chlamydomonas reinhardtii nuclear gene SIM30 suppresses translational defects caused by initiation codon mutations in chloroplast genes. Genetics 145:935–943
Choquet Y, Zito F, Wostrikoff K, Wollman FA (2003) Cytochrome f translation in Chlamydomonas chloroplast is autoregulated by its carboxyl-terminal domain. Plant Cell 15:1443–1454
Cosner ME, Jansen RK, Palmer JD, Downie SR (1997) The highly rearranged chloroplast genome of Trachelium caeruleum (Campanulaceae): multiple inversions, inverted repeat expansion and contraction, transposition, insertions/deletions, and several repeat families. Curr Genet 31:419–429
Cui X, Hsia AP, Liu F, Ashlock DA, Wise RP, Schnable PS (2003) Alternative transcription initiation sites and polyadenylation sites are recruited during Mu suppression at the rf2a locus of maize. Genetics 163:685–698
Drager RG, Zeidler M, Simpson CL, Stern DB (1996) A chloroplast transcript lacking the 3′ inverted repeat is degraded by 3′→5′ exoribonuclease activity. RNA 2:652–663
Drager RG, Girard-Bascou J, Choquet Y, Kindle KL, Stern DB (1998) In vivo evidence for 5′→3′ exoribonuclease degradation of an unstable chloroplast mRNA. Plant J 13:85–96
Eibl C, Zou Z, Beck A, Minkyun K, Mullet J, Koop H-U (1999) In vivo analysis of plastid psbA, rbcL and rpl32 UTR elements by chloroplast transformation: tobacco plastid gene expression is controlled by modulation of transcript levels and translation efficiency. Plant J 19:333–345
Felder S, et al (2001) The nucleus-encoded HCF107 gene of Arabidopsis provides a link between intercistronic RNA processing and the accumulation of translation-competent psbH transcripts in chloroplasts. Plant Cell 13:2127–2141
Harris EH (1989) The Chlamydomonas sourcebook: a comprehensive guide to biology and laboratory use. Academic Press, San Diego
Hayes R, Kudla J, Schuster G, Gabay L, Maliga P, Gruissem W (1996) Chloroplast mRNA 3′-end processing by a high molecular weight protein complex is regulated by nuclear encoded RNA binding proteins. EMBO J 15:1132–1141
Higgs DC, Shapiro RS, Kindle KL, Stern DB (1999) Small cis-acting sequences that specify secondary structures in a chloroplast mRNA are essential for RNA stability and translation. Mol Cell Biol 19:8479–8491
Kuras R, Wollman F-A (1994) The assembly of cytochrome b 6 /f complexes: an approach using genetic transformation of the green alga Chlamydomonas reinhardtii. EMBO J 13:1019–1027
Lee H, Bingham SE, Webber AN (1996) Function of 3′ non-coding sequences and stop codon usage in expression of the chloroplast psaB gene in Chlamydomonas reinhardtii. Plant Mol Biol 31:337–354
Legendre M, Gautheret D (2003) Sequence determinants in human polyadenylation site selection. BMC Genomics 4:7
Levy H, Kindle KL, Stern DB (1997) A nuclear mutation that affects the 3′ processing of several mRNAs in Chlamydomonas chloroplasts. Plant Cell 9:825–836
Maniatis T, Fritsch EM, Sambroook J (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Press, Cold Spring Harbor, N.Y.
Matsumoto T, Matsuo M, Matsuda Y (1991) Structural analysis and expression during dark–light transitions of a gene for cytochrome f in Chlamydomonas reinhardtii. Plant Cell Physiol 32:863–872
Maul JE, et al (2002) The Chlamydomonas reinhardtii plastid chromosome: islands of genes in a sea of repeats. Plant Cell 14:2659–2679
Memon AR, Meng B, Mullet JE (1996) RNA-binding proteins of 37/38 kDa bind specifically to the barley chloroplast psbA 3′-end untranslated RNA. Plant Mol Biol 30:1195–1205
Monde RA, Greene JC, Stern DB (2000a) The sequence and secondary structure of the 3′-UTR affect 3′-end maturation, RNA accumulation, and translation in tobacco chloroplasts. Plant Mol Biol 44:529–542
Monde RA, Schuster G, Stern DB (2000b) Processing and degradation of chloroplast mRNA. Biochimie 82:573–582
Morton BR, Clegg MT (1993) A chloroplast DNA mutational hotspot and gene conversion in a noncoding region near rbcL in the grass family (Poaceae). Curr Genet 24:357–365
Newman SM, Harris EH, Johnson AM, Boynton JE, Gillham NW (1992) Nonrandom distribution of chloroplast recombination events in Chlamydomonas reinhardtii: evidence for a hotspot and an adjacent cold region. Genetics 132:413–429
Palmer JD (1991) Plastid chromosomes: structure and evolution. In: Vasil IK (ed) The molecular biology of plastids. Academic Press, San Diego
Palmer JD, Boynton JE, Gillham NW, Harris EH (1985) Evolution and recombination of the large inverted repeat in Chlamydomonas chloroplast DNA. In: Arntzen C, Bogorad L, Bonitz S, Steinback K (eds) Molecular biology of the photosynthetic apparatus. Cold Spring Harbor Press, Cold Spring Harbor, N.Y., pp 269–278
Rott R, Drager RG, Stern DB, Schuster G (1996) The 3′ untranslated regions of chloroplast genes in Chlamydomonas reinhardtii do not serve as efficient transcriptional terminators. Mol Gen Genet 252:676–683
Rott R, Levy H, Drager RG, Stern DB, Schuster G (1998a) 3′-processed mRNA is preferentially translated in Chlamydomonas reinhardtii chloroplasts. Mol Cell Biol 18:4605–4611
Rott R, Liveanu V, Drager RG, Stern DB, Schuster G (1998b) The sequence and structure of the 3′-untranslated regions of chloroplast transcripts are important determinants of mRNA accumulation and stability. Plant Mol Biol 36:307–314
Sakamoto W, Kindle KL, Stern DB (1993) In vivo analysis of Chlamydomonas chloroplast petD gene expression using stable transformation of β-glucuronidase translational fusions. Proc Natl Acad Sci USA 90:497–501
Sakamoto W, Sturm NR, Kindle KL, Stern DB (1994) petD mRNA maturation in Chlamydomonas reinhardtii chloroplasts: The role of 5′ endonucleolytic processing. Mol Cell Biol 14:6180–6186
Schuster G, Gruissem W (1991) Chloroplast mRNA 3′ end processing requires a nuclear-encoded RNA-binding protein. EMBO J 10:1493–1502
Schwartz S et al. (2000) PipMaker—a web server for aligning two genomic DNA sequences. Genome Res 10:577–586
Shepherd HS, Boynton JE, Gillham NW (1979) Mutations in nine chloroplast loci of Chlamydomonas affecting photosynthetic functions. Proc Natl Acad Sci USA 76:1353–1357
Staub JM, Maliga P (1994) Translation of psbA mRNA is regulated by light via the 5′-untranslated region in tobacco plastids. Plant J 6:547–553
Stern DB, Gruissem W (1987) Control of plastid gene expression: 3′ inverted repeats act as mRNA processing and stabilizing elements, but do not terminate transcription. Cell 51:1145–1157
Stern DB, Kindle KL (1993) 3′ end maturation of the Chlamydomonas reinhardtii chloroplast atpB mRNA is a two-step process. Mol Cell Biol 13:2277–2285
Stern DB, Radwanski ER, Kindle KL (1991) A 3′ stem/loop structure of the Chlamydomonas chloroplast atpB gene regulates mRNA accumulation in vivo. Plant Cell 3:285–297
Sturm N, et al (1994) The petD gene is transcribed by functionally redundant promoters in Chlamydomonas reinhardtii chloroplasts. Mol Cell Biol 14:6171–6179
Thompson RJ, Mosig G (1987) Stimulation of a Chlamydomonas chloroplast promoter by novobiocin in situ and in E. coli implies regulation by torsional stress in the chloroplast DNA. Cell 48:281–287
Walter M, Kilian J, Kudla J (2002) PNPase activity determines the efficiency of mRNA 3′-end processing, the degradation of tRNA and the extent of polyadenylation in chloroplasts. EMBO J 21:6905–6914
Zerges W (2000) Translation in chloroplasts. Biochimie 82:583–601
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415
Acknowledgements
We thank Linda Rymarquis and Katia Wostrikoff for their assistance in RNA and protein analysis, Elise Kikis in preparing PCR probes for S1 protection, and Jude Maul for SDR analysis. This work was supported by National Science Foundation award MCB 0091020 to D.B.S., with early experiments supported by a Georges Morel Prize and Guggenheim Fellowship awarded to D.B.S. That phase of the work was carried out in the laboratory of Francis-André Wollman, Institut de Biochimie Physico-Chimique, Paris, where D.B.S. was a sabbatical visitor. We thank all members of that laboratory for their interest and support. C.L.S. was supported by an Olin Graduate Fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F.-A. Wollman
Rights and permissions
About this article
Cite this article
Jiao, H.S., Hicks, A., Simpson, C. et al. Short dispersed repeats in the Chlamydomonas chloroplast genome are collocated with sites for mRNA 3′ end formation. Curr Genet 45, 311–322 (2004). https://doi.org/10.1007/s00294-004-0487-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-004-0487-7