
Abstract
Using uidA (β-glucuronidase; GUS) reporter gene constructs, the 5′-untranslated region (UTR) of the Chlamydomonas chloroplast rbcL gene was screened by deletion and mutational analysis for the presence of a promoter element that previous studies implied to reside within the first 63 base pairs of the UTR. Deleting a large segment of the rbcL 5′UTR in a 3′→5′ direction to position +36, changing the remaining 36 base pairs at the 5′ end of the UTR, and increasing by five base pairs the distance between the rbcL 5′UTR and the basic promoter element located at position −10 did not abolish transcription from the basic rbcL promoter. It is concluded that the apparent loss of transcriptional activity found in earlier studies after deletion of sequences downstream of the transcription initiation site is due to the synthesis of very unstable transcripts that escape detection by Northern analysis and in vivo transcription assays. Chimeric rbcL:GUS transcripts containing changes in the beginning of the 5′UTR that affect RNA secondary structure are estimated to be at least 50 times less stable than rbcL:GUS transcripts containing the non-modified rbcL 5′UTR sequence.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The molecular machinery that controls and regulates gene expression in plastids is basically prokaryotic. Reflecting the bacterial ancestry of plastids are, for instance, the structures of plastid RNA polymerases (Maliga 1998), the sequences of plastid gene promoters (Igloi and Kössel 1992), prokaryotic ribosome-binding sites (Hirose et al. 1998), and the structures of other elements, like stem-loops at the 3′ ends of plastid transcripts (Rott et al. 1998). Understanding the functioning of these features is imperative for future work that aims at inserting and expressing foreign DNA in plastid genomes (Heifetz 2000).
We previously analyzed and described the structures of two types of promoter in the chloroplast genome of the unicellular green alga Chlamydomonas reinhardtii: a promoter resembling the typical Escherichia coli sigma 70 promoter, comprising −10 (TATAAT) and −35 (TTGACA) consensus sequence elements, and a promoter that lacks the −35 element but has an extended −10 (TATAATAT) sequence (Klein et al. 1992). The latter seems to be the common promoter of protein genes in the Chlamydomonas chloroplast, whereas the sigma 70-like promoter—which appears to be more active than the common promoter of protein genes—is found in front of the ribosomal RNA genes in Chlamydomonas. Sigma 70-like promoters and promoters that lack −10 and −35 consensus sequences have also been identified in the genomes of plastids in higher plants (Igloi and Kössel 1992; Hajdukiewicz et al. 1997; Silhavy and Maliga 1998; Sriraman et al. 1998).
Based on deletion analysis of the promoter region of the Chlamydomonas chloroplast rbcL gene, which showed a loss of transcriptional activity when sequences beyond position +63 (the start site of transcription is taken as +1) were deleted in a 3′→5′ direction, we inferred that, in addition to the −10 element, the common protein gene promoter comprises sequences downstream of the transcription start site between positions +1 and +63 (Klein et al. 1994). This study aimed at delineating and defining the sequence of the presumptive downstream promoter element. The results of the analysis suggested, however, that the extended −10 sequence alone is sufficient to direct transcription in the chloroplast of C. reinhardtii. The apparent loss of transcriptional activity after a 3′→5′ deletion beyond position +63 is in fact due to the synthesis of extremely unstable transcripts that escape detection by in vivo transcription assays. A rough calculation of the extent of transcript destabilization shows that transcript degradation is enhanced at least 50-fold by changing sequences in the rbcL 5′ untranslated region (UTR).
Materials and methods
Algal strains and cultures
The DNA constructs described below were transformed into the non-photosynthetic atpB gene-deficient mutant ac-uc-2-21 mt+ (CC-373) obtained from the Chlamydomonas Genetics Center at Duke University, N.C. The mutant was grown at room temperature in dim light (<0.05 μmol sec−1 m−2) in high-salt (HS) medium (Sueoka 1960) supplemented with 2.5 g potassium acetate l−1. Chloroplast transformants were grown in HS medium in high-light conditions (50 μmol sec−1 m−2) either as 100-ml cultures at room temperature in 250-ml Erlenmeyer flasks or as 150-ml cultures at 32 °C in glass tubes in a water bath. The latter cultures were continuously bubbled with 2% CO2-enriched air and diluted each day approximately 5-fold with fresh HS medium. For isolation of total RNA and in vivo transcription assays, cultures were grown in 12 h light/12 h dark cycles. Cells for RNA isolation and transcription assays were taken at 11 h in the dark period.
Chloroplast transformation
Mutant ac-uc-2-21 was transformed by bombardment with DNA-coated 1.0-μm tungsten or 0.6-μm gold particles using a PDS-1000/He system (BIORAD, Hercules, Calif.) and following protocols described by Boynton et al. (1988) and Blowers et al. (1989). All DNA constructs were cloned into the transformation vector pCrc32 (Blowers et al. 1993). Upon transformation, chloroplast DNA sequences missing in the mutant were complemented by sequences contained in the DNA of the transformation vector (Blowers et al. 1989), thereby restoring the photosynthetic capacity of the cells. Transformants were selected by growth on HS medium in high-light conditions.
Plasmids
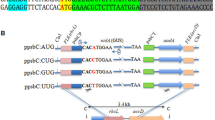
The 3′→5′ deletion sequences (+15, +16, +36, +55; Fig. 1B) of the 5′UTR of the rbcL gene were synthesized by PCR, using a 5′ primer complementary to sequences around position −70 (Fig. 1A) and four 3′ primers complementary to sequences around positions +15, +26, +36, and +55 of the rbcL 5′UTR (Fig. 1). To facilitate cloning of the PCR products, the four 3′ primers were designed with an SspI restriction site at their 3′ ends. PCR fragments were digested with XhoI and SspI and cloned into XhoI/EcoRV-digested pBluescript SK+ (Stratagene, La Jolla, Calif.). The inserts were released by digesting the plasmids with XhoI/SmaI and were cloned in front of the uidA (β-glucuronidase; GUS) reporter gene into the XhoI/SmaI-cut transformation vector pCrc32. The 3′→5′ deletion to position +45 (Fig. 1B) was constructed by digesting the +55 PCR product with BspEI (BspEI cuts at position +41 in the rbcL 5′UTR; Fig. 1A, B) and blunting the 3′ end with the Klenow fragment of E. coli DNA polymerase I before digestion with XhoI. The fragment was cloned into pBluescript SK+ and pCrc32, as described above for the other PCR fragments. All other rbcL 5′UTR sequences (Fig. 1C, D) were synthesized full-length as complementary oligonucleotides with a blunt 5′ end and a BspEI-compatible 3′ end. The fragments were cloned into the SwaI/BspEI sites of vector +157/SK+, which harbors a rbcL:GUS construct (rbcL sequences from positions −70 to +157; Fig. 1A) cloned into the XhoI/XbaI sites of pBluescript SK+ (Anthonisen et al. 2002). The rbcL:GUS inserts were released from +157/SK+ by digestion with XhoI/XbaI and cloned into the XhoI/XbaI-digested transformation vector pCrc32.

Chimeric rbcL:β-glucuronidase (GUS) constructs used in this work. A Schematic drawing of the 5′ region of the rbcL gene cloned in front of the GUS-coding region in rbcL:GUS constructs. Numbers above the drawing indicate the position relative to the start site of transcription (+1). The rbcL 5′ untranslated region (UTR) is marked as a black box (positions +1 to +92), the promoter sequence (P) is shown as a gray box, and the enhancer-like sequence (E) is indicated as a striped box. Restriction sites used for cloning are indicated. B The sequence of the first 63 bp of the rbcL 5′UTR and the progressive 3′→5′ deletions analyzed in this work. Lengths of the DNA segments remaining after deletion are indicated below the sequence. Numbers at the ends of the segments specify the position of the terminal 3′ nucleotide of the remaining 5′UTR sequence. A previously identified RNA-stabilizing sequence element (positions +38 to +47; Anthonisen et al. 2001) is underlined. C The four sequences of the rbcL 5′UTR (positions +1 to +46) in which segments of 9 bp were changed. Changes cover the region from positions +1 to +36. The altered sequences are underlined. They all contain a SphI site (GCATGC) used for easy identification of desired clones in plasmid preparations. D Sequences of the rbcL 5′UTR in which all base pairs between positions +6 and +36 (p6-36) or +5 and +37 (p5-37) are changed. In sequence p6-36, pyrimidine nucleotides were changed to pyrimidines (T to C, or C to T) or purine nucleotides to purines (A to G). In p5-37, the complete sequence from position 5 to 37 was inserted in reverse orientation. Transcripts from these sequences are predicted to form the same stem-loop structure at their 5′ ends as the original rbcL 5′UTR sequence (Fig. 4A). Sequence p+5 (at the bottom) has an additional five base pairs inserted between the rbcL 5′UTR and upstream rbcL promoter sequences (indicated by small letters). At the RNA level, the insertion results in five extra nucleotides at the 5′ terminus of rbcL:GUS transcripts
Southern and Northern analyses
Genomic DNA and total RNA were isolated from chloroplast transformants as described by Blowers et al. (1989) and Salvador et al. (1993a). Genomic DNA was screened for the presence of the GUS gene by slot-blot hybridizations or DNA gel (Southern) blots on samples digested with KpnI and HindIII. Slot blots and Northern blots were hybridized to the entire coding region of the GUS gene (approx. 1.9 kb), random primer-labeled with 32P-alpha dCTP (Amersham Biosciences, Buckinghamshire, UK). Southern blots were hybridized to the 700-bp HpaI-EcoRV restriction fragment from plasmid pCrcatpB containing a portion of the coding region of the Chlamydomonas chloroplast atpB gene (Blowers et al. 1990). Hybridization and washing conditions were as described by Church and Gilbert (1984).
In vivo determination of transcription rates
Relative rates of transcription of the chimeric rbcL:GUS genes, the endogenous rbcL gene, and the endogenous atpB gene were determined in the dark by measuring the incorporation of 32P-labeled inorganic phosphate into GUS transcripts as described by Baker et al. (1984) and Blowers et al. (1990). The intensity of hybridization signals on autoradiograms was determined with 1-D image analysis software (Kodak Digital Science, Rochester, N.Y.). Ratios of transcription rates shown in Figs. 2, 3, and 4 were calculated from the increase in signal intensity at 10 min and 20 min, corrected for the lengths of the transcripts, and normalized to the signal intensity of transcripts of the endogenous rbcL gene.

Analysis of 3′→5′ deletion constructs in Chlamydomonas chloroplast transformants. A GUS transcript levels in transformants carrying 3′→5′ deletions, as determined by RNA gel blot (Northern) analysis. Numbers above the lanes indicate the position of the 3′ terminal nucleotide in the rbcL 5′UTR (see Fig. 1B). Results are from the same autoradiogram (overnight exposure with intensifying screen at −80 °C), but the figure is composed of lanes that were originally separated on the RNA gel. B Determination of relative rates of transcription of the chimeric GUS gene, compared with the endogenous atpB and rbcL genes. Numbers below the autoradiograms indicate the positions of the 3′ terminal nucleotides of the rbcL 5′UTR in the 3′→5′ deletion constructs. The control shows the rates of transcription of a construct in which the complete rbcL 5′ region from positions −70 to +157 (see Fig. 1A) is fused to the GUS-coding region. Normalized ratios of the rates of gene transcription calculated from the increase in signal intensity between the 10-min and 20-min time-points are shown below the autoradiograms. Kodak 1-D image analysis software was used for the quantitative analysis of signal intensities. Values were corrected for the size of each transcripts (rbcL 1.6 kb, atpB 1.9 kb, rbcL:GUS 2.4 kb)

Analysis of GUS gene expression in transformants harboring chimeric rbcL:GUS genes in which 9-bp segments in the rbcL 5′UTR were altered. A Northern analysis of RNA isolated from transformants carrying the four constructs shown in Fig. 1C. Numbers above the lanes denote the constructs as shown in Fig. 1C. B Relative rates of GUS gene transcription in transformants carrying the p1-9 or p10-18 constructs. The control is the same as in Fig. 2B. The ratios of relative rates of transcription shown below the autoradiograms were calculated as explained in Fig. 2B

Predicted RNA conformations at the 5′ ends of transcripts from wild-type and mutated rbcL:GUS genes, RNA gel blot (Northern) analysis of total RNA, and the determination of relative rates of gene transcription in Chlamydomonas chloroplast transformants harboring rbcL:GUS genes containing the changes in the rbcL 5′UTR specified in Fig. 1D. A Secondary structures at the 5′ ends of wild-type and mutated 5′UTRs, as predicted by the mfold program (Zuker et al. 1999). The sequence previously shown to function as an RNA stabilizing element is boxed (Anthonisen et al. 2001). B Autoradiogram of a Northern analysis of RNA isolated from Chlamydomonas chloroplast transformants harboring rbcL:GUS constructs containing the 5′UTR mutations shown in Fig. 1D. C Relative rates of GUS gene transcription in transformants carrying a rbcL:GUS gene, into which five base pairs (small letters) were inserted between the rbcL 5′UTR and upstream promoter sequences. A previously delineated promoter element in the −10 region is boxed. Bent arrows below the sequences denote the start site and direction of transcription. Signal intensities and ratios were analyzed as explained in Fig. 2B. WT Wild-type DNA sequence
Results
Sequences between positions +1 and +36 of the rbcL 5′UTR are required for transcription or transcript stability
The basic promoter (TATAATAT) of the rbcL gene in the chloroplast genome of C. reinhardtii is located about ten nucleotides upstream of the start site of transcription (Fig. 1A; Klein et al. 1992). Transcription from this promoter has been found to be augmented by an enhancer-like sequence element positioned in the beginning of the rbcL coding region, between nucleotides +108 and +143 relative to the start site of transcription (Fig. 1A; Klein et al. 1994; Anthonisen et al. 2002). Based on the analysis of 3′→5′ deletions, which abolished transcription upon deleting nucleotides between positions +63 and +1 (Klein et al. 1994), an additional promoter element is postulated to reside within the first 63 bp of the rbcL 5′UTR. To delineate the 3′ border of the putative promoter element, we constructed a number of chimeric rbcL:GUS genes with progressive 3′→5′ deletions into the 5′UTR portion of the rbcL:GUS genes to positions +55, +45, +36, +26, and +15 (Fig. 1B).
GUS transcripts accumulate to normal levels in transformants harboring 3′→5′ rbcL UTR deletion constructs up to position +55 (Fig. 2A) but cannot be detected by Northern analysis in transformants harboring deletions +45 and beyond (Fig. 2A). In vivo transcription assays showed that chimeric GUS genes are transcribed in transformants carrying +45 or +36 rbcL UTR deletions, albeit at relatively low levels, while transcription of the chimeric GUS reporter gene drops below 10% of the transcription rate of the rbcL gene when sequences between positions +36 and +26 and beyond are deleted (Fig. 2B). Deletion constructs +45 and +36 are apparently transcribed at only approx. 20% of the rate of the control gene. It is possible, though, that the transcription rates of these constructs are actually higher because disruption of a RNA-stabilizing sequence element located at positions +38 to +47 (Fig. 1B; Fig. 4A; Anthonisen et al. 2001) might render transcripts unstable and result in less accumulation in the transcription assay. Nevertheless, the results strongly suggested that the putative promoter element is located between positions +1 and +36 of the rbcL 5′UTR.
Changing all nucleotides between positions +1 and +36 does not abolish GUS transcription
To find the putative promoter element, the first 36 nucleotides of the rbcL 5′UTR were mutated in four rbcL:GUS constructs, each with a block of mutations from positions +1 to +9, +10 to +18, +19 to +27, and +28 to +36, respectively (Fig. 1C). Only transformants harboring the +19 to +27 mutations accumulated GUS transcripts at easily detectable levels (Fig. 3A) suggesting that either transcription of the other rbcL:GUS genes is severely impaired or the GUS transcripts in these transformants are unstable. Measuring rates of GUS transcription in transformants carrying the rbcL:GUS genes with the +1 to +9 and +10 to +18 mutations revealed that both genes are in fact transcribed, although apparently at lower rates than a control gene without mutations in the 5′UTR segment (Fig. 3B). Note that the transcription rate of the +28 to +36 mutant construct was not measured because, by the time we got the transformant, the analysis of transformants carrying the constructs shown in Fig. 1D (Fig. 4) showed that the +28 to +36 sequence did not contain the putative promoter element.
The results suggested that transcripts from rbcL:GUS genes in which segments +1 to +9 or +10 to +18 are mutated are unstable and do not accumulate to levels that are readily detectable by RNA gel (Northern) blot analysis. However, the transcripts are still stable enough to accumulate to levels that can be detected in vivo by the transcription assay.
The 3′→5′ deletions into the rbcL 5′UTR render transcripts susceptible to rapid degradation
The in vivo transcription assay measures the accumulation of [32P]-labeled transcripts after exposure of phosphate-starved cells to [32P]-labeled inorganic phosphate for 10 min and 20 min (Baker et al. 1984). Therefore, if half-lives of transcripts to be studied are well below 5 min [decay rates follow an exponential curve and depend on the kinetics of the nucleolytic enzyme(s)], the assay will indicate no transcription, even when transcripts are synthesized at normal rates but do not accumulate to detectable levels. It appears conceivable that the apparent loss of transcription upon deleting the rbcL 5′UTR beyond position +36 in a 3′→5′ direction (Fig. 2B) could in fact be due to a sharp decline in transcript stability (the half-life of rbcL:GUS transcripts has been determined to be around 4–5 h in the dark; Salvador et al. 1993b) and not to a lack of transcript synthesis. To test this notion, we tried to mutate the region between positions +1 and +36 in the rbcL 5′UTR without affecting transcript stability.
A rbcL:GUS gene was constructed in which almost all nucleotides in the +1 to +36 region were altered but the structural and sequence elements suspected to be important for transcript stability were preserved. Previous work (Anthonisen et al. 2001) showed the presence of a stem-loop structure and a transcript-stabilizing sequence (Fig. 4A) in the beginning of the rbcL 5′UTR. Preserving both, we replaced all adenine nucleotides in the region between positions +6 and +36 by guanine nucleotides and all thymine nucleotides by cytosine nucleotides, and vice versa (construct p6-36 in Fig. 1D). The new gene had a completely different sequence in the beginning of the 5′UTR, except for the first five nucleotides. The five nucleotides were preserved because they were expected to be important for transcript stability, due to base pairing with nucleotides of an RNA-stabilizing sequence element located at positions +38 to +47 (Anthonisen et al. 2001; Fig. 4A). The nucleotides were, however, already altered in the p1-9 construct without abolishing transcription (Fig. 1C, Fig. 3B).
Transcripts synthesized from the new gene (p6-36) could form the same stem-loop structure at the 5′ end, but with a different sequence from the original rbcL 5′UTR (Fig. 4A). Chloroplast transformants harboring the p6-36 rbcL:GUS gene accumulated GUS transcripts to easily detectable levels (Fig. 4B), showing that the gene is transcribed and thus showing that an essential promoter element is not present in the +6 to +36 region of the rbcL 5′UTR.
It is possible, though, that swapping all adenine and thymine nucleotides with guanine and cytosine nucleotides, respectively, strongly affects transcription but that the increased number of hydrogen bonds in the now G/C-rich 5′ stem-loop structure stabilizes the transcripts to an extent that allows the accumulation of transcripts over time, even at extremely low rates of transcription. Another rbcL:GUS gene was constructed in which the DNA region between positions +5 and +37 was inverted (p5-37; Fig. 1D). The DNA sequence from positions +5 to +37 in the resulting gene differed significantly from the corresponding sequence in the original rbcL 5′UTR (and from the sequence in the p6-36 UTR) but, when transcribed, the resulting transcripts were predicted to form the same original stem-loop structure at their 5′ ends with the same number of hydrogen bonds as the native rbcL transcripts (Fig. 4A). Transformants carrying this construct accumulated GUS transcripts to easily detectable levels (p5-37 in Fig. 4B), indicating that the gene is transcribed and again showing the lack of an essential promoter element in the rbcL 5′UTR between positions +5 and +37.
Additional support for the notion of a lack of an essential promoter element in the rbcL 5′UTR comes from the analysis of Chlamydomonas transformants carrying rbcL:GUS constructs in which the distance between the rbcL 5′UTR and upstream DNA sequences is changed (Fig. 4C). The spacing between promoter elements is shown to be critical for promoter strength, probably because optimal spacing facilitates an open complex formation or stability after binding the RNA polymerase adjacent to the transcription initiation site (Stefano and Gralla 1982). For example, the activity of sigma 70-type E. coli promoters is very sensitive to changes in the length of the spacer sequence between the −10 and −35 regions (Aoyama et al. 1983), an addition of two extra base pairs leading to an 85% decrease in promoter activity (Mandecki and Reznikoff 1982). The insertion of five base pairs between the rbcL 5′UTR and upstream DNA sequences (Fig. 4C, construct p+5), which increased the length of the spacer between the −10 promoter region and a putative promoter element downstream in the rbcL 5′UTR to an extent that could completely inactivate the E. coli sigma 70-type promoter, did not abolish transcription of the GUS gene in Chlamydomonas transformants (Fig. 4C). Rates of GUS gene transcription that are higher than the transcription rates from the endogenous atpB gene promoter (Fig. 4C) show that the rbcL promoter in the insertion construct is functioning and that there is no essential promoter element downstream of the transcription initiation site in the Chlamydomonas rbcL gene. Furthermore, because the five extra nucleotides significantly alter the nucleotide sequence at the transcription initiation site (Fig. 4C), the results strongly suggest that those nucleotides are not crucial for transcription initiation.
Despite relatively high rates of GUS gene transcription in transformants harboring the p+5 construct (Fig. 4C), GUS transcripts do not accumulate in the transformants to levels detectable by RNA gel blot (Northern) assays (Fig. 4B). Evidently, the addition of the five extra nucleotides to the 5′ terminus of the rbcL 5′UTR must render the transcripts susceptible to rapid degradation. Considering this instability of the rbcL:GUS transcripts carrying five extra nucleotides at their 5′ termini, we believe that the rates of rbcL:GUS gene transcription shown in Fig. 4C are actually higher than those determined by the transcriptional assay, but that the assay is affected by concurrent degradation of rbcL:GUS transcripts during the RNA-sampling period.
Taken together, the failure to delineate by various approaches a DNA sequence in the rbcL 5′UTR that is essential for transcription suggests that earlier results indicating the presence of a promoter element in this region were biased, due to the rapid degradation of transcripts after deleting sequence elements crucial for transcript stability. Considering that the half-life of transcripts from control rbcL:GUS genes is around 4–5 h (Salvador et al. 1993b), compared with an estimated half-life of well below 5 min for a number of transcripts with mutated rbcL 5′UTRs, the RNA-stabilizing elements in the 5′UTR sequence must increase the longevity of transcripts at least 50-fold.
Discussion
The results of this study point to a dilemma inherent in determinations of rates of transcription in vivo. The assays fail to detect even normal rates of transcription when transcripts are extremely unstable and do not accumulate to detectable levels. In our in vivo transcription assay, the first sample is usually taken 10 min after the addition of labeled phosphate. Transcripts with a half-life well below 5 min are thus not seen by the assay. Because the half-life of unmodified rbcL:GUS transcripts is around 4–5 h (Salvador et al. 1993b), we initially considered it unlikely that the sequence modifications and deletions we introduced into the rbcL 5′UTR could accelerate degradation to an extent that would interfere with the in vivo transcription assay. However, provided essential RNA stability elements were preserved (Fig. 4), it was possible to change all nucleotides of the rbcL sequence, in which an essential promoter element was suspected to be located, without abolishing transcription. These data, together with the result that spacing between the rbcL 5′UTR and the −10 promoter region is not critical for basic promoter activity (Fig. 4C), support the conclusion that the previously measured loss of transcriptional activity in 3′→5′ deletion constructs is in fact due to rapid degradation of 5′-modified transcripts and not to lack of transcription.
It is generally assumed that the average stability of transcripts in different species is linked to the cell cycle period of that species, i.e. the shorter the cell cycle, the shorter the half-lives of transcripts. This could allow organisms to adjust the composition of their RNA pool (transcriptome) when conditions change due to environmental or internal cues. For example, E. coli transcripts have been found to have relatively short half-lives (in the range of a few minutes; Bouvet and Belasco 1992) compared with transcripts in cyanobacteria (e.g. Kujat and Owttrim 2000) or eukaryotic organisms (e.g. Albig and Decker 2001). Chlamydomonas chloroplast transcripts have been found to be stable for several hours (Salvador et al. 1993a). The fact that the longevity of transcripts in the Chlamydomonas chloroplast can drop from hours to less than a few minutes suggests that these transcripts are normally protected from degradation and that they are rapidly degraded only when the protection is removed.
Although this study did not aim at identifying elements in the rbcL 5′UTR sequence that are important for RNA stability, the results inadvertently show (Fig. 4) that most, but not all, of the sequences at the 5′ end of the rbcL 5′UTR are irrelevant for transcript stability, at least in dark conditions. In this respect, our data confirm previous reports (Drager et al. 1999; Higgs et al. 1999; Nickelsen et al. 1999; Anthonisen et al. 2001) showing that cis-acting sequences and structural features at the 5′ ends of chloroplast transcripts play a significant role in transcript accumulation, in particular that stem-loop structures predicted to form in the 5′ regions of a number of chloroplast transcripts might be essential for protection against nucleolytic degradation (Higgs et al. 1999; Zou et al. 2003). A previously identified cis-acting sequence element in the rbcL 5′UTR, which was shown to be crucial for transcript stability (Anthonisen et al. 2001), is partly included in the predicted stem-loop structure of the rbcL 5′UTR and might thus function as a binding site for a protective trans-acting factor. It is tempting to speculate that such trans-acting factors could be related to or be identical with nuclear-encoded proteins that are required for the stability of some Chlamydomonas chloroplast RNAs (Boudreau et al. 2000; Nickelsen 2000, 2003; Vaistij et al. 2000), but direct evidence for the binding of these proteins to RNA-stabilizing sequences is missing.
Chloroplast promoters of higher plants can be grouped into at least two classes: (1) bacterial sigma 70-type promoters containing −10 and −35 consensus sequences and (2) promoters with a loose consensus sequence near the transcription initiation site (Igloi and Kössel 1992; Hajdukiewicz et al. 1997; Silhavy and Maliga 1998). Variants of the latter type of promoter have been found (e.g. Sriraman et al. 1998). The sigma 70-like promoter appears to be exclusively recognized by a plastid-encoded bacterium-type RNA polymerase (PEP), while a nucleus-encoded phage-type RNA polymerase (NEP) transcribes genes from the other type of promoter (Maliga 1998; Hess and Börner 1999). Inactivation of one of the RNA polymerases does not prevent plant growth, because a number of genes in the chloroplasts of higher plants appear to be transcribed by both types of RNA polymerase (Hajdukiewicz et al. 1997). In Chlamydomonas, rpo genes encoding a PEP are present in the chloroplast genome, but a NEP homologue has not yet been detected in Chlamydomonas (Lilly et al. 2002). Thus, genes in the Chlamydomonas chloroplast appear to be transcribed exclusively by the PEP protein complex, which thus recognizes at least two types of promoter, the sigma 70-type and a promoter with an extended −10 region. The rbcL gene promoter is an example of the latter type of promoter. It is likely that, as in bacteria, specific binding of the PEP to each type of promoter is mediated by sigma-like proteins.
References
Albig AR, Decker CJ (2001) The target of rapamycin signaling pathway regulates mRNA turnover in the yeast Saccharomyces cerevisiae. Mol Biol Cell 12:3428–3438
Anthonisen IL, Salvador ML, Klein U (2001) Specific sequence elements in the 5′ untranslated regions of rbcL and atpB gene mRNAs stabilize transcripts in the chloroplast of Chlamydomonas reinhardtii. RNA 7:1024–1033
Anthonisen IL, Kasai S, Kato K, Salvador ML, Klein U (2002) Structural and functional characterization of a transcription-enhancing sequence element in the rbcL gene of the Chlamydomonas chloroplast genome. Curr Genet 41:349–356
Aoyama T, Takanami M, Ohtsuka E, Taniyama Y, Marumoto R, Sato H, Ikehara M (1983) Essential structure of E. coli promoter: effect of spacer length between the two consensus sequences on promoter function. Nucleic Acids Res 11:5855–5864
Baker EJ, Schloss JA, Rosenbaum JL (1984) Rapid changes in tubulin RNA synthesis and stability induced by deflagellation in Chlamydomonas. J Cell Biol 99:2074–2081
Blowers AD, Bogorad L, Shark KB, Sanford JC (1989) Studies on Chlamydomonas chloroplast transformation: foreign DNA can be stably maintained in the chromosome. Plant Cell 1:123–132
Blowers AD, Ellmore GS, Klein U: Bogorad L (1990) Transcriptional analysis of endogenous and foreign genes in chloroplast transformants of Chlamydomonas. Plant Cell 2:1059–1070
Blowers AD, Klein U, Ellmore GS, Bogorad L (1993) Functional in vivo analyses of the 3′ flanking sequences of the Chlamydomonas rbcL and psaB genes. Mol Gen Genet 238:339–349
Boudreau E, Nickelsen J, Lemaire SD, Ossenbühl F, Rochaix J-D (2000) The Nac2 gene of Chlamydomonas encodes a chloroplast TPR-like protein involved in psbD mRNA stability. EMBO J 19:3366–3376
Bouvet P, Belasco JG (1992) Control of RNase E-mediated RNA degradation by 5′-terminal base pairing in E. coli. Nature 360:488–491
Boynton JE, Gillham NW, Harris EH, Hosler JP, Johnson AM, Jones AR, Randolph-Anderson BL, Robertson D, Klein TM, Shark KB, Sanford JC (1988) Chloroplast transformation in Chlamydomonas with high velocity microprojectiles. Science 240:1534–1538
Church G, Gilbert W (1984) Genomic sequencing. Proc Natl Acad Sci USA 8:1991–1995
Drager RG, Higgs DC, Kindle KL, Stern DB (1999) 5′ to 3′ exoribonucleolytic activity is a normal component of chloroplast mRNA decay pathways. Plant J 19:521–531
Hajdukiewicz PTJ, Allison LA, Maliga P (1997) The two RNA polymerases encoded by the nuclear and the plastid compartments transcribe distinct groups of genes in tobacco plastids. EMBO J 16:4041–4048
Heifetz PB (2000) Genetic engineering of the chloroplast. Biochimie 82:655–666
Hess WR, Börner T (1999) Organellar RNA polymerases of higher plants. Int Rev Cytol 190:1–59
Higgs DC, Shapiro RS, Kindle KL, Stern DB (1999) Small cis-acting sequences that specify secondary structures in a chloroplast mRNA are essential for RNA stability and translation. Mol Cell Biol 19:8479–8491
Hirose T, Kusumegi T, Sugiura M (1998) Translation of tobacco chloroplast rps14 mRNA depends on a Shine–Dalgarno-like sequence in the 5′-untranslated region but not on internal RNA editing in the coding region. FEBS Lett 430:257–260
Igloi GL, Kössel H (1992) The transcriptional apparatus of chloroplasts. Crit Rev Plant Sci 10:525–558
Klein U, Camp JD de, Bogorad L (1992) Two types of chloroplast gene promoters in Chlamydomonas reinhardtii. Proc Natl Acad Sci USA 89:3453–3457
Klein U, Salvador ML, Bogorad L (1994) Activity of the Chlamydomonas chloroplast rbcL gene promoter is enhanced by a remote sequence element. Proc Natl Acad Sci USA 91:10819–10823
Kujat SL, Owttrim GW (2000) Redox-regulated RNA helicase expression. Plant Physiol 124:703–714
Lilly JW, Maul JE, Stern DB (2002) The Chlamydomonas reinhardtii organellar genomes respond transcriptionally and post-transcriptionally to abiotic stimuli. Plant Cell 14:2681–2706
Maliga P (1998) Two plastid RNA polymerases of higher plants: an evolving story. Trends Plant Sci 3:4–6
Mandecki W, Reznikoff WS (1982) A lac promoter with a changed distance between −10 and −35 regions. Nucleic Acids Res 11:903–912
Nickelsen J (2000) Mutations at three different nuclear loci of Chlamydomonas suppress a defect in chloroplast psbD mRNA accumulation. Curr Genet 37:136–142
Nickelsen J (2003) Chloroplast RNA-binding proteins. Curr Genet 43:392–399
Nickelsen J, Fleischmann M, Boudreau E, Rahire M, Rochaix J-D (1999) Identification of cis-acting RNA leader elements required for chloroplast psbD gene expression in Chlamydomonas. Plant Cell 11:957–970
Rott R, Levy H, Drager RG, Stern DB, Schuster G (1998) The sequence and structure of the 3′-untranslated regions of chloroplast transcripts are important determinants of mRNA accumulation and stability. Plant Mol Biol 36:307–314
Salvador ML, Klein U, Bogorad L (1993a) Light-regulated and endogenous fluctuations of chloroplast transcript levels in Chlamydomonas. Regulation by transcription and RNA degradation. Plant J 3:213–219
Salvador ML, Klein U, Bogorad L (1993b) 5′ sequences are important positive and negative determinants of the longevity of Chlamydomonas chloroplast gene transcripts. Proc Natl Acad Sci USA 90:1556–1560
Silhavy D, Maliga P (1998) Mapping of promoters for the nucleus-encoded plastid RNA polymerase (NEP) in the iojap maize mutant. Curr Genet 33:340–344
Sriraman P, Silhavy D, Maliga P (1998) The phage-type PclpP-53 plastid promoter comprises sequences downstream of the transcription initiation site. Nucleic Acids Res 26:4874–4879
Stefano JE, Gralla JD (1982) Spacer mutations in the lac ps promoter. Proc Natl Acad Sci USA 79:1069–1072
Sueoka N (1960) Mitotic replication of deoxyribonucleic acid in Chlamydomonas reinhardi. Proc Natl Acad Sci USA 46:83–91
Vaistij FE, Boudreau E, Lemaire SD, Goldschmidt-Clermont M, Rochaix J-D (2000) Characterization of Mbb1, a nucleus-encoded tetratricopeptide-like repeat protein required for expression of the chloroplast psbB/psbT/psbH gene cluster in Chlamydomonas reinhardtii. Proc Natl Acad Sci USA 97:14813–14818
Zou Z, Eibl C, Koop H-U (2003) The stem-loop region of the tobacco psbA 5′UTR is an important determinant of mRNA stability and translation efficiency. Mol Gen Genomics 269:340–349
Zuker M, Mathews DH, Turner DH (1999) Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide. In: Barciszewski J, Clark BFC (eds) RNA biochemistry and biotechnology. (NATO ASI Series) Kluwer, Dordrecht, pp 11–43
Acknowledgements
Parts of this work were supported by grants PB98-1445 and BMC2003-0329 from the Ministerio de Ciencia y Tecnologia to M.L.S. and grant 100946-/410 from the Norwegian Research Council to U.K.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F.-A. Wollman
Rights and permissions
About this article
Cite this article
Salvador, M.L., Suay, L., Anthonisen, I.L. et al. Changes in the 5′-untranslated region of the rbcL gene accelerate transcript degradation more than 50-fold in the chloroplast of Chlamydomonas reinhardtii . Curr Genet 45, 176–182 (2004). https://doi.org/10.1007/s00294-003-0470-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-003-0470-8