
Abstract
A gene encoding a trypsin protease was isolated from a tomato isolate of Verticillium dahliae. The gene, designated VTP1, contains two introns and is predicted to encode a protein of 256 amino acids. The gene is present in V. dahliae isolates from different host plants and in V. albo-atrum; weakly hybridizing sequences are present in V. tricorpus. VTP1 cDNA sequences were identified in a sequence tag analysis of genes expressed under growth conditions that promote microsclerotia development. Replacement of the gene, by Agrobacterium tumefaciens-mediated transformation (ATMT), with a mutant allele construct did not noticeably alter either pathogenicity or growth in culture. Searches of expressed sequence tag databases showed that, in addition to the VTP1 gene, V. dahliae contains two genes encoding subtilisin-like proteases similar to those produced by pathogenic Aspergillus spp. This is the first description of the application of ATMT to the molecular analysis of phytopathogenic Verticillium spp.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Verticillium dahliae Kleb. causes vascular wilt in a wide variety of dicotyledonous plant species, and produces a variety of extracellular hydrolases, most notably pectinases and cellulases, which have long been considered important for the induction of disease symptoms (for a review, see Pegg 1981; Pegg and Brady 2002; Puhalla and Bell 1981). Protease activity has been reported in V. dahliae (Dobinson et al. 1997; Lambert and Pujarniscle 1984; St. Leger et al. 1997), which is able to hydrolyze a broad range of complex proteinaceous substrates.
Proteases have been proposed to facilitate penetration of the plant cell walls by fungal pathogens, degrade host defense-related proteins, and/or to allow the fungus to utilize complex plant cell wall proteins as a nutrient source during colonization of the plant (Dobinson et al. 1997; Murphy and Walton 1996). However, studies of the foliar pathogens Cochliobolus carbonum and Stagonospora nodorum and the vascular wilt fungus Fusarium oxysporum f. sp. lycopersici have demonstrated that targeted mutation of individual protease genes in these species does not affect overall growth or pathogenicity (Bindschedler et al. 2003; DiPietro et al. 2001; Murphy and Walton 1996). Moreover, Murphy and Walton (1996) identified in C. carbonum both a trypsin and a subtilisin and demonstrated that mutation of the trypsin gene resulted in only a 35–45% reduction in total protease activity, while in F. oxysporum, mutation of a subtilase gene had no significant effect on overall levels of protease activity (DiPietro et al. 2001). Similarly, Bindschedler et al. (2003) found that the SNP1 trypsin accounted for only approximately 50% of the protease activity in a wild-type strain of S. nodorum and that, in the absence of that enzyme, a subtilisin-like protease could be detected.
It has been reported that the phytopathogenic Verticillium species lack subtilisins (Bidochka et al. 1999; Segers et al. 1999). However, St. Leger et al. (1997) described low levels of subtilisin activity in both V. dahliae and V. albo-atrum isolates for which trypsin-like enzymes were the primary proteases. Given that these species are poor saprophytes in field soil (Schnathorst 1981) and grow more slowly in culture than the more aggressive wilt pathogen F. oxysporum f. sp. lycopersici, we were interested in determining whether a previously identified extracellular trypsin protease from V. dahliae (Dobinson et al. 1997) is functionally redundant, as appears to be the case with the F. oxysporum f. sp. lycopersici extracellular proteases (DiPietro et al. 2001). We report here the cloning and mutation, by Agrobacterium tumefaciens-mediated transformation (ATMT), of the gene encoding the V. dahliae trypsin, and a preliminary analysis of gene sequences corresponding to two subtilisin-like proteases that were identified during the course of an expressed sequence tag (EST) analysis of V. dahliae.
Despite the abundance of molecular methods that have been developed for the study of fungal plant pathogens and the worldwide economic importance of verticillium wilt, this disease is poorly represented in the arena of molecular plant pathology. The results of our experiments demonstrate that targeted gene disruptions may be readily produced in V. dahliae by transposon mutagenesis of a cloned gene of interest, followed by transfer of the mutant gene into V. dahliae via ATMT. These tools, together with the ability to search recently generated EST databases (Neumann and Dobinson 2003) for genes of interest, thus provide us with valuable resources for the molecular analysis of phytopathogenic Verticillium spp.
Materials and methods
Fungal strains and growth conditions
All isolates of V. dahliae and other Verticillium spp used in this study are derived from single-spore cultures and maintained as described by Dobinson (1995). Cultures were grown on complete medium (CM; Dobinson et al. 1997) agar and in liquid CM unless otherwise specified. Hygromycin B-resistant strains were grown on CM agar amended with 25 μg hygromycin B/ml. Spore inoculum was prepared from cultures grown in liquid CM, as described by Dobinson et al. (1997).
Nucleic acid manipulations
Genomic DNA used for DNA blot hybridizations was purified from V. dahliae spores using a glass bead breakage method (Dobinson 1995). Genomic DNA used for library construction was purified from nuclei of V. dahliae isolate Dvd-T5 (ATCC accession 201177), as described by Timberlake (1986). Plasmids were purified from bacterial cultures by an alkaline lysis method using the QIAprep 8 miniprep kit (Qiagen, Mississauga, Canada).
A lambda genomic DNA library was constructed from Dvd-T5 DNA partially digested with Sau3AI and cloned into λGEM11 XhoI half-site arms (Promega Corp., Madison, Wis.), using procedures recommended by the supplier. The library was screened by plaque hybridization with the 41-base oligonucleotide 5′-ACTTTGAAGGGAAAGTCGCCGGCGCTGGCAGCAACACCACC-3′, which was designed based on the N-terminal amino acid sequence of the V. dahliae trypsin VDP30 (Dobinson et al. 1997) and the codon usage of a F. oxysporum trypsin gene (Rypniewski et al. 1993). The oligonucleotide was 5′ end-labeled with [γ32P]-ATP, using Ready-To-Go T4 polynucleotide kinase (Amersham Pharmacia Biotech, Baie d’Urfé, Canada). Hybridizations and washes were done at 50 °C, as described by Dobinson et al. (1993).
A plasmid library of HindIII fragments (1.8–2.3 kb) from Dvd-T5 nuclear DNA was constructed in pBluescript II KS (+); the HindIII-digested DNA was fractionated by electrophoresis through 0.8% low-melting-point agarose gels and recovered from the agarose by treatment with β-agarase (New England Biolabs, Mississauga, Canada), using conditions recommended by the supplier. The library was screened by colony hybridization with a digoxigenin (DIG)-labeled PCR product (probe II, see Fig. 2), amplified as described below. These and all other hybridizations using DIG-labelled probes were done as described by Dobinson et al. (1998), except that salmon sperm DNA (final concentration 50 μg/ml) was added to the prehybridization and hybridization buffers.
For DNA blot hybridizations, genomic DNA (approx. 500 ng) was digested with the restriction enzymes specified, fractionated through 0.8% agarose gels, and transferred by capillary blotting to Hybond N+ membranes (Amersham Pharmacia Biotech).
DNA sequence analysis was done at the John P. Robarts Research Institute Sequencing Facility (London, Canada) or at the Southern Crop Protection and Food Research Centre sequencing facility at Agriculture and Agri-Food Canada (London, Canada), as described by Neumann and Dobinson (2003), using custom primers and primers complementary to cloning site sequences in the plasmid vectors. Protein sequences were aligned using the ClustalW algorithm of MegaAlign (DNAStar, Madison, Wis.), and shaded using the publicly available program Boxshade ver. 3.21.
To construct the VTP1 gene-replacement vector, a 2-kb HindIII fragment containing the full-length gene and flanking sequences was subcloned into the binary vector pDHt (Mullins et al. 2001). The cloned gene was subjected to in vitro transposon mutagenesis, using the EZ::TN system (Epicentre Technologies, Madison, Wis.) and custom vector pSK846, which is derived from pMOD-2 (Epicentre Technologies) and contains, between the EZ::TN transposase recognition sequences, the chloramphenicol resistance gene amplified from pBCSK (Stratagene, La Jolla, Calif.) and the hygromycin B resistance gene cassette from plasmid pCB1004 (Carroll et al. 1994). The transposon was released from pSK846 by digestion with XmnI. Mutagenized plasmids were propagated in Escherichia coli strain DH5α MRF′ and chloramphenicol-resistant clones that contained the transposon inserted into the VTP1 gene coding region were identified by PCR amplification, as described below, using primers complementary to sequences at the 5′ and 3′ ends of the VTP1 coding sequence. Transposon insertion sites within the coding region were determined by sequence analysis, as described above, using the pMOD-2 forward and reverse sequencing primers (Epicentre Technologies).
PCR amplification reactions contained 1× PCR buffer (20 mM Tris-HCl, pH 8.4, 50 mM KCl), 2.5 mM MgCl2, 200 μM of each dNTP, 5–7 pmol of each primer, 0.5 units of Taq or Platinum Taq DNA polymerase (Invitrogen, Burlington, Canada), and template (1–5 ng of plasmid, 10–15 ng of genomic DNA, or approx. 8×105 plaque-forming units of phage stock from a developing microsclerotia cDNA library; Neumann and Dobinson 2003). A denaturation step (2 min at 94 °C) was followed by 30 cycles of denaturation (45 s at 94 °C), annealing (45 s at the specified temperatures), and elongation (1 min at 72 °C), followed by a final elongation step (5–7 min at 72 °C). DIG-labelled amplicons used for DNA blot hybridizations were synthesized from plasmid templates, using the PCR-DIG labeling kit (Roche Diagnostics, Laval, Canada), primers complementary to vector sequences flanking the cloning site, and an annealing temperature of 65 °C.
For RT-PCR analyses, fungal cultures were grown on basal medium agar and total RNA was extracted with TRIzol reagent (Invitrogen), as described by Neumann and Dobinson (2003). Prior to use, RNA (0.5 μg in sterile distilled water) was treated with amplification grade DNAse I (Invitrogen) and reverse transcribed from an oligo(dT)12–18 primer with SuperScript II Rnase H− reverse transcriptase (Invitrogen), according to the supplier’s protocols. VTP1 and actin gene sequences were amplified from the cDNA and genomic DNA as described above, using an annealing temperature of 60 °C. Reactions contained both VTP1 gene-specific primers (5′-GCGGTGGCTGGTTCCTATCAAC-3′ and 5′-CAACGACTTCGCCATCTGGAAG-3′) and actin gene-specific primers (5′-GCCCTCTTCCAGCCCTCCGTTCTC-3′ and 5′-TCGGCGTGGTTTTGTGGTGAG-3′); both primer sets flanked introns in the respective genes.
Transformation-mediated mutagenesis of V. dahliae VTP1 gene
ATMT of V. dahliae was done as described by Mullins et al. (2001), with several modifications as follows: (1) prior to co-cultivation, V. dahliae was grown in liquid CM and conidia were harvested as described above, (2) A. tumefaciens AGL-1 cells containing the gene-replacement vector were grown in induction medium without acetosyringone, and (3) A. tumefaciens cells and V. dahliae spores were co-cultivated for 2 days on a nitrocellulose membrane, before the membrane was transferred to selective medium (CM containing 50 μg hygromycin B/ml, 100 μg moxalactam/ml, 200 μM cefotaxime). Hygromycin B-resistant transformants were single-spore purified as described by Dobinson (1995) and screened by PCR amplification and DNA blot hybridization, as described above, to identify gene-replacement mutants.
Protease activity and pathogenicity assays
Protease activity was assessed by growing cultures on a skim-milk agar medium (Dobinson et al. 1997) containing 2% or 1% skim-milk powder and 0.5% glucose. Cultures were incubated at 24 °C or 28 °C for 12 days.
Pathogenicity was assessed using 14-day-old tomato seedlings (cv. Bonny Best) and a root dip inoculation method, as described by Dobinson et al. (1996).
Results
Characterization of the V. dahliae VTP1 gene
DNA sequences corresponding to an extracellular trypsin-like protease (previously designated VDP30) produced by the V. dahliae tomato isolate Dvd-T5 (Dobinson et al. 1997) were isolated by screening the lambda Dvd-T5 genomic DNA library with an oligonucleotide probe, as described in the Materials and methods. Under the low stringency conditions of the hybridization, the probe hybridized to six families of clones. DNA sequence analysis indicated that only one of these families contained trypsin gene sequences. The full-length gene (designated VTP1) (GenBank accession AY354459), located on a 2-kb HindIII fragment, was subsequently isolated from a plasmid library of HindIII restriction fragments and sequenced in both directions, as described in the Materials and methods.
The VTP1 sequence encodes a predicted amino acid sequence of 256 amino acids (28.2 kDa); the nucleotide sequence surrounding the putative translation initiation codon (CATCATGGTT) is consistent with the fungal translation initiation consensus sequence (Edelmann and Staben 1994). Comparison of the genomic DNA sequence with that of the cDNA (amplified from recombinant phage of a V. dahliae cDNA library, as described in the Materials and methods) confirmed the presence of two small introns (56 bp, 66 bp) with 5′ and 3′ donor sites and splice branch sites similar to those in previously characterized fungal introns (Edelmann and Staben 1994). A polyadenylation site 307 bp downstream of the translation stop codon was identified by sequence analysis of a clone from the same cDNA library.
DNA blot hybridization analysis showed that VTP1 gene sequences are highly conserved in the genomes of V. dahliae strains isolated from a variety of host plants, including tomato, potato, and Japanese maple, and in isolates of the closely related plant pathogen V. albo-atrum (Fig. 1). Weakly hybridizing sequences were also detected in isolates of another plant-associated species, V. tricorpus, suggesting that this species contains a VTP1 homologue, but none were detected in V. nigrescens or in the mushroom pathogen V. fungicola (Fig. 1).

Southern hybridization analysis of the VTP1 gene in Verticillium spp. Genomic DNA was digested with XhoI and hybridized at 65 °C with the digoxigenin (DIG)-labeled VTP1 gene probe I (shown in Fig. 2), as described in the Materials and methods. V. dahliae strains from tomato (Dvd-T5, Dvd-2), eggplant (Dvd-E6), and Japanese maple (VdJM), V. nigrescens isolate from eggplant (Dvn-E1), V. tricorpus (Vt1, Vt2), V. fungicola (1659), V. albo-atrum (w2). Sizes of DIG-labeled molecular size markers (Roche Molecular Biochemicals) are indicated to the right of the figure. Equivalent amounts of genomic DNA from each Verticillium isolate were in each lane, as indicated by ethidium bromide staining of the gel (data not shown)
Transformation-mediated disruption of the VTP1 gene
For gene disruption analysis, the HindIII fragment carrying the VTP1 gene was subcloned into the binary vector pDHt and subjected to in vitro transposon mutagenesis. A mutant plasmid (pEATn30) was selected in which the transposon was inserted after the 13th base of the second intron (Fig. 2A). A V. dahliae VTP1 gene-replacement mutant was generated by ATMT of the mutant gene into strain Dvd-T5. Among the first few transformants analyzed, one (VDAT6-6) was identified in which the wild-type gene had been replaced by the mutant allele (Fig. 2B). Although transcription of VTP1 was abolished in this transformant (Fig. 2C), its growth rate on a variety of media was not significantly different from that of the wild-type strain, nor were microsclerotia production or pathogenicity noticeably affected (data not shown).

Targeted mutation of the V. dahliae VTP1 gene. A VTP1 gene disruption vector pEATn30. Black boxes VTP1 gene coding region, white boxes introns. The insertion site of the approx. 2.5-kb transposon containing the hygromycin B resistance gene cassette is indicated by an arrowhead beneath the second intron. Restriction sites: B BamHI, H HindIII, S SacI, X XhoI. The locations of hybridization probes used in this study are indicated by the open boxes (I, II, III) below the restriction map. B Southern blot analysis of wild-type strain Dvd-T5 and hygromycin B-resistant transformants (VDAT6-1 to VDAT6-7) containing the VTP1 gene disruption construct. Genomic DNA was digested with BamHI and hybridized with DIG-labeled VTP1 gene probe III, as described in the Materials and methods. The asterisk denotes the gene-replacement mutant, and the arrow indicates the restriction fragment (approx. 360 bp) that spans the BamHI sites in the transposon and the VTP1 open reading frame. Sizes of selected DIG-labeled molecular weight markers are indicated to the right of the photograph. C RT-PCR analysis of Dvd-T5 and VDAT6-6, grown for 7 days on basal agar medium. RT-PCR was done as described in the Materials and methods, using VTP1 gene-specific primers (Vtp1) and actin gene-specific primers (A). The VTP1 and actin gene amplicons generated from the cDNA are approx. 450 bp and 400 bp, respectively. PCR amplification from Dvd-T5 genomic DNA (gDNA) yielded a product of approx. 500 bp for each primer set. Sizes of molecular size markers in lane M are indicated to the left of the photograph
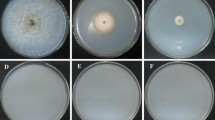
When V. dahliae is grown on a skim-milk agar medium, protease production is detected as a zone of clearing around the colonies. At 28 °C, growth of all strains on medium containing either 1% or 2% skim-milk was much slower than at 24 °C. Under all growth conditions, the zone of clearing initially appeared to be somewhat reduced in VDAT6-6, in comparison with that of Dvd-T5 and the ectopic transformants (data not shown). However, within 12 days post-inoculation, no consistent differences were seen between the strains (Fig. 3, data not shown).

Growth and protease production of the wild-type strain Dvd-T5, gene disruption mutant VDAT6-6, and ectopic transformants VDAT6-3 and VDAT6-7. Cultures were grown for 12 days at 24 °C on an agar medium containing 2% skim-milk, as described in the Materials and methods
Identification of two subtilase family protease genes in V. dahliae
During the course of an EST analysis of V. dahliae (Neumann and Dobinson 2003), cDNA clones were identified that corresponded to VTP1 and to two genes encoding putative extracellular proteases not previously identified in V. dahliae. Sequence comparisons showed that the encoded proteins (VSP1, VSP2) were similar to members of the peptidase S8 family of subtilases, including the Aspergillus fumigatus oryzin (54% identity with VSP1, 51% with VSP2), and A. oryzae oryzin (50% identity with VSP1, 49% with VSP2) and contained the conserved aspartate, histidine, and serine residues of the subtilase catalytic triad (Fig. 4). Although the VSP1 gene sequence was first identified in the cDNA library generated from V. dahliae cells grown under conditions that promote near-synchronous development of microsclerotia, and the VSP2 sequence was identified in the library generated from cells grown in a simulated xylem fluid medium (Neumann and Dobinson 2003), the two sequences could be amplified from both cDNA libraries (data not shown).

Alignment of translated cDNA sequences from VSP1 and VSP2 (GenBank accessions AY356397 and AY356398, respectively) with subtilase family enzymes Aspergillus oryzae (Ao) oryzin (accession P12547), and A. fumigatus (Af) oryzin (accession P28296). Alignment was done as described in the Materials and methods. Identical amino acids are highlighted in black and indicated in the consensus line with asterisks. Conserved amino acids are highlighted in gray and indicated in the consensus line with dots. The amino acids of the catalytic triad are indicated with asterisks above the sequences
Discussion
We cloned and characterized the VTP1 gene corresponding to the previously identified VDP30 trypsin-like protease from V. dahliae (Dobinson et al. 1997). Comparison of the experimentally determined amino-terminal sequence from purified VDP30 with that of the predicted VTP1 protein sequence showed that the purified protein lacked the 29 N-terminal amino acids of the predicted protein. These data indicate that, as with other trypsins, the primary translation product of the VTP1 gene undergoes proteolytic cleavage to release the mature protein. Although amino acid 1 and amino acid 15 of VDP30 (Dobinson et al. 1997) differed from those in the predicted sequence encoded by VTP1, such discrepancies are not uncommon and have been attributed to protein sequencing errors (Görlach et al. 1998; Scott-Craig et al. 1992).
The results of the gene knockout (KO) experiment demonstrated that the trypsin encoded by VTP1 is not essential for overall growth and pathogenicity. The gene disruption data also indicated that V. dahliae produces at least one other protease and, in our expressed sequence tag databases (Neumann and Dobinson 2003), we identified genes corresponding to two proteases in the S8 subtilase family. These data are thus consistent with the results of similar studies on the proteases of other foliar or root-invading phytopathogenic fungi (Bindschedler et al. 2003; DiPietro et al. 2001; Murphy and Walton 1996) and with the overall level of redundancy among extracellular hydrolases produced by plant pathogenic Verticillium spp (Bidochka et al. 1999; St. Leger et al. 1997). Bindschedler et al. (2003) suggested that the loss of a single protease in S. nodorum could be compensated for by the production of additional proteases. That this may also occur with V. dahliae is supported by our finding that, although subtilisin activity has either not been detected in trypsin-producing isolates of V. dahliae and V. albo-atrum (Bidochka et al. 1999; Segers et al. 1999), or been detected at only low levels (St. Leger et al. 1997), V. dahliae strain Dvd-T5 expresses at least two subtilisin-like genes when grown in vitro under conditions designed to mimic in planta nutrient conditions.
Trypsin proteases may be particularly well suited to the degradation of plant proteins and may indeed have been co-opted by plant-pathogenic Verticillium species for that purpose, as has been suggested by St. Leger et al. (1997). However, although the limited saprophytic capacity of these species (Schnathorst 1981) suggested to us that the previously identified V. dahliae trypsin (Dobinson et al. 1997) might be important for in planta growth, it appears that, as with the more aggressive wilt pathogen F. oxysporum (DiPietro et al. 2001), V. dahliae maintains an array of proteases for the degradation of complex, proteinaceous nutrient sources.
Since the first report of DNA transfer into Saccharomyces cerevisiae by Agrobacterium tumefaciens (Bundock et al. 1995), ATMT has been successfully applied to a diverse array of fungi (Abuodeh et al. 2000; Chen et al. 2000; Combier et al. 2003; Covert et al. 2001; de Groot et al. 1998; Gouka et al. 1999; Hanif et al. 2002; Malonek and Meinhardt 2001; Mullins et al. 2001; Rho et al. 2001; Sullivan et al. 2002; Zhang et al. 2003; Zwiers and De Waard 2001.). ATMT exhibits a number of advantages as a method for transforming fungi, including: (1) high efficiency transformation (de Groot et al. 1998; Mullins et al. 2001; Rho et al. 2001), (2) increased frequency of homologous recombination (Bundock et al. 1999; Mullins et al. 2001; Zwiers and De Waard 2001), (3) the ability to transform spores, hyphae, and even mushroom fruiting body tissue, thus circumventing the problems (low yield, viability) typically associated with protoplast isolation (Chen et al. 2000; de Groot et al. 1998; Mullins et al. 2001), and (4) low copy number of inserted T-DNA per genome (de Groot et al. 1998; Mullins et al. 2001; Rho et al. 2001; Sullivan et al. 2002). To our knowledge, this is the first report of the use of ATMT for transformation and gene disruption in V. dahliae. To facilitate the rapid generation of mutant alleles for gene KO via ATMT, in this study we also tested an in vitro transposon-mediated mutagenesis technique. A conventional method for constructing a mutant allele is to replace parts of a target gene with an antibiotic resistance gene using unique restriction enzyme sites, which is time-consuming and often limits the size of gene fragments that can be used. Based on the data in this report and our ability to use a combination of transposon tagging and ATMT for the mutagenesis of several other genes (Klimes and Dobinson 2003; Kang et al., unpublished data; Dobinson et al., unpublished data), this gene KO method provides us with a valuable tool for the molecular analysis of verticillium wilt.
References
Abuodeh RO, Orbach MJ, Mandel MA, Das A, Galgiani JN (2000) Genetic transformation of Coccidioides immitis facilitated by Agrobacterium tumefaciens. J Infect Dis 181:2106–2110
Bidochka MJ, Burke S, Ng L (1999) Extracellular hydrolytic enzymes in the fungal genus Verticillium: adaptations for pathogenesis. Can J Microbiol 45:856–864
Bindschedler LV, Sanchez P, Dunn S, Mikan J, Thangavelu M, Clarkson JM, Cooper RM (2003) Deletion of the SNP1 trypsin protease from Stagonospora nodorum reveals another major protease expressed during infection. Fungal Genet Biol 38:43–53
Bundock P, Dulk-Ras A den, Beijersbergen A, Hooykaas PJJ (1995) Trans-kingdom T-DNA transfer from Agrobacterium tumefaciens to Saccharomyces cerevisiae. EMBO J 14:3206–3214
Carroll AM, Sweigard JA, Valent B (1994) Improved vectors for selecting resistance to hygromycin. Fungal Genet Newsl 41:22
Chen X, Stone M, Schlagnhaufer C, Romaine CP (2000) A fruiting body tissue method for efficient Agrobacterium-mediated transformation of Agaricus bisporus. Appl Environ Microbiol 66:4510–4513
Combier JP, Melayah D, Raffier C, Gay G, Marmeisse R (2003) Agrobacetrium tumefaciens-mediated transformation as a tool for insertional mutagenesis in the symbiotic ectomycorrhizal fungus Hebeloma cylindrosporum. FEMS Microbiol 220:141–148
Covert SF, Kapoor P, Lee M, Briley A, Nairn CJ (2001) Agrobacterium tumefaciens-mediated transformation of Fusarium circinatum. Mycol Res 105:259–264
DiPietro A, Huetas-Gonzalez MD, AGutierrez-Corona JF, Martinez-Cadena G, Meglecz E, Roncero MIG (2001) Molecular characterization of a subtilase from the vascular wilt fungus Fusarium oxysporum. Mol Plant-Microbe Interact 14:653–662
Dobinson KF (1995) Genetic transformation of the vascular wilt fungus Verticillium dahliae. Can J Bot 73:710–715
Dobinson KF, Harris RE, Hamer JE (1993) Grasshopper, a long terminal repeat (LTR) retroelement in the phytopathogenic fungus Magnaporthe grisea. Mol Plant-Microbe Interact 6:114–126
Dobinson KF, Tenuta GK, Lazarovits G (1996) Occurrence of race 2 of Verticillium dahliae in processing tomato fields in southwestern Ontario. Can J Plant Pathol 18:55–58
Dobinson KF, Lecomte N, Lazarovits G (1997) Production of an extracellular trypsin-like protease by the fungal plant pathogen Verticillium dahliae. Can J Microbiol 43:227–233
Dobinson KF, Patterson NA, White GJ, Grant S (1998) DNA fingerprinting and vegetative compatibility analysis indicate multiple origins for Verticillium dahliae race 2 tomato isolates from Ontario, Canada. Mycol Res 102:1089–1095
Edelmann SE, Staben C (1994) A statistical analysis of sequence features within genes from Neurospora crassa. Exp Mycol 18:70–81
Görlach JM, Van Der Knaap E, Walton JD (1998) Cloning and targeted disruption of MLG1, a gene encoding two of three extracellular mixed-link glucanases of Cochliobolus carbonum. Appl Env Microbiol 64:385–391
Gouka RJ, Gerk C, Hooykaas PJJ, Bundock R, Musters W, Verrips CT, Groot MJA de (1999) Transformation of Aspergillus awamori by Agrobacterium tumefaciens-mediated homologous recombination. Nat Biotechnol 17:598–601
Groot MJA de, Bundock P, Hooykaas PJJ, Beijersbergen AGM (1998) Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat Biotechnol 16:839–842
Hanif M, Pardo AG, Gorfer M, Raudaskoski M (2002) T-DNA transfer and integration in the ectomyrrhizal fungus Suillus bovinus using hygromycin B as a selectable marker. Curr Genet 41:183–188
Klimes A, Dobinson KF (2003) Functional characterization of a Verticillium dahliae hydrophobin gene homologue. Can J Plant Pathol 25:321
Lambert F, Pujarniscle S (1984) Purification and properties of the proteinase produced in vitro by Verticillium dahliae. Can J Microbiol 30:1488–1493
Malonek S, Meinhardt F (2001) Agrobacterium tumefaciens-mediated genetic transformation of the phytopathogenic ascomycete Calonectria morganii. Curr Genet 40:152–155
Mullins ED, Chen X, Romaine P, Raina R, Geiser DM, Kang S (2001) Agrobacterium-mediated transformation of Fusarium oxysporum: an efficient tool for insertional mutagenesis and gene transfer. Phytopathology 91:173–180
Murphy JM, Walton JD (1996) Three extracellular proteases from Cochliobolus carbonum: cloning and targeted disruption of ALP1. Mol Plant-Microbe Interact 9:290–297
Neumann MJ, Dobinson KF (2003) Sequence tag analysis of gene expression during pathogenic growth and microsclerotia development in the vascular wilt pathogen Verticillium dahliae. Fungal Genet Biol 38:54–62
Pegg GF (1981) Biochemistry and physiology of pathogenesis. In: Mace ME, Bell AA, Beckman CH (eds) Fungal wilt diseases of plants. Academic Press, New York, pp 193–253
Pegg GF, Brady BL (2002) Verticillium wilts. CABI, Wallingford
Puhalla JE, Bell AA (1981) Genetics and biochemistry of wilt pathogens. In: Mace ME, Bell AA, Beckman CH (eds) Fungal wilt diseases of plants. Academic Press, New York, pp 146–192
Rho HS, Kang S, Lee YH (2001) Agrobacterium tumefaciens-mediated transformation of the plant pathogenic fungus, Magnaporthe grisea. Mol Cells 12:407–411
Rypniewski WR, Hastrup S, Betzel C, Dauter M, Dauter Z, Papendorf G, Branner S, Wilson KS (1993) The sequence and X-ray structure of the trypsin from Fusarium oxysporum. Protein Eng 6:341–348
Schnathorst WC (1981) Life cycle and epidemiology of Verticillium. In: Mace ME, Bell AA, Beckman CH (eds) Fungal wilt diseases of plants. Academic Press, New York, pp 81–111
Scott-Craig JS, Panaccione DG, Pocard J-A, Walton JD (1992) The cyclic peptide synthetase catalyzing HC-toxin production in the filamentous fungus Cochliobolus carbonum is encoded by a 15.7-kilobase open reading frame. J Biol Chem 267:26044–26049
Segers R, Butt TM, Carder JH, Keen JN, Kerry BR, Peberdy JF (1999) The subtilisins of fungal pathogens of insects, nematodes and plants: distribution and variation. Mycol Res 103:395–402
St. Leger RJ, Joshi L, Roberts DW (1997) Adaptation of proteases and carbohydrases of saprophytic, phytopathogenic and entomopathogenic fungi to the requirements of their ecological niches. Microbiology 143:1983–1992
Sullivan TD, Rooney PJ, Klein BS (2002) Agrobacterium tumefaciens integrates transfer DNA into single chromosomal sites of dimorphic fungi and yields homokaryotic progeny from multinucleate yeast. Eukaryot Cell 1:895–905
Timberlake WE (1986) Isolation of stage- and cell-specific genes from fungi. In: Bailey J (ed) Biology and molecular biology of plant–pathogen interactions, vol 1. Springer, Berlin Heidelberg New York, pp 343–357
Zhang A, Lu P, Dahl-Roshak AM, Paress PS, Kennedy S, Tkacz JS, An Z (2003) Efficient disruption of a polyketide synthase gene (pks1) required for melanine synthesis through Agrobacterium-mediated transformation of Glarea lozoyensis. Mol Gen Genomics 268:645–655
Zwiers LH, De Waard MA (2001) Efficient Agrobacterium tumefaciens-mediated gene disruption in the phytopathogen Mycosphaerella graminicola. Curr Genet 39:388–393
Acknowledgements
We thank E. Antone for technical assistance, I. van Grinsven and S. Millar for sequence analysis, A. Molnar for preparation of Figs. 3A and 4, S. Evans for assistance with preparation of other figures, and M. Neumann for critical reading of the manuscript. This study was supported in part by funding from the Natural Sciences and Engineering Research Council of Canada.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Heitman
Rights and permissions
About this article
Cite this article
Dobinson, K.F., Grant, S.J. & Kang, S. Cloning and targeted disruption, via Agrobacterium tumefaciens-mediated transformation, of a trypsin protease gene from the vascular wilt fungus Verticillium dahliae . Curr Genet 45, 104–110 (2004). https://doi.org/10.1007/s00294-003-0464-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-003-0464-6