
Abstract.
The genetic transformation of plastids of higher plants has developed into a powerful approach for both basic research and biotechnology. Due to the high copy number of the plastid genome per plastid and per cell, repeated cycles of shoot regeneration under conditions selective for the modified plastid chromosome are required to obtain transformants entirely lacking wild-type plastid genomes. The presence of promiscuous plastid DNA in nuclear and/or mitochondrial genomes that generally contaminate even gradient-purified plastid fractions reduces the applicability of the highly sensitive PCR approach to monitor the absence of residual wild-type plastid chromosomes in transformed lines. It is therefore difficult, or even impossible, to assess reliably the hetero- or homoplastomic state of plastid transformants in this manner. By analysing wild-type and transplastomic mutants of tobacco, we demonstrate that separation of plastid chromosomes isolated from gradient-purified plastid fractions by pulsed-field gel electrophoresis can overcome the problem of (co)amplification of interfering promiscuous plastid DNA. PCR analyses with primers specific for plastid, mitochondrial and nuclear genes reveal an impressive purity of such plastid DNA fractions at a detection limit of less than one wild-type plastid chromosome copy per ten transplastomic cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genetic alteration of plastid genomes (plastomes) has become an important tool not only for basic but also for applied research. It provides access to plastid gene function and expression and to the structure–function relationships of plastome-encoded components and processes. Because of various advantages over transformation of the nuclear genome, notably the targeted integration of transformed DNA by homologous recombination, the high expression rates of introduced genes, the possibility of simultaneous expression of multiple transgenes from a single operon (transgene stacking) and the virtual lack of transgene pollen transmission in most plants, plastid transformation has an increasing impact on biotechnology (for a review, see Bogorad 2000; Bock 2001; Daniell et al. 2002; Maliga 2002). The targeted transformation of plastomes can be obtained by biolistic and polyethylene glycol-based approaches or by agitating cells with glass beads in the presence of the transformation vector (Boynton et al. 1988; Svab et al. 1990; Kindle et al. 1991; Golds et al. 1993; Svab and Maliga 1993; Koop and Kofer 1995). The former two methods are also applicable to higher plants. Although a growing number of species is becoming amenable to plastid transformation (Sikdar et al. 1998; Khan and Maliga 1999; Sidorov et al. 1999; Ruf et al. 2001), it was hitherto routinely used with only the unicellular green alga Chlamydomonas reinhardtii and tobacco.
The genetic potential of plastids is deposited in a circular double-stranded DNA molecule, generally in the size range 120–250 kbp, depending on the organism. This corresponds to some 100–250 genes, including a few highly conserved reading frames (ycfs, hypothetical chloroplast reading frames), for which functions are still unknown. The availability of plastid transformation technologies allows one to experimentally unravel the role of such ycfs by a reverse genetic approach. Generally, the gene of interest is replaced or interrupted by a marker gene that is flanked by regions of a few hundred base pairs with identity to the plastome target site required for homologous recombination. Regeneration of plants under conditions selective for marker gene expression then leads to transplastomic plants. Due to the approximately 50–100 chloroplasts per tobacco leaf cell and between 50 and 100 chromosome copies per chloroplast (cf. Herrmann and Possingham 1980; Bendich 1987), the primary transformed material is heteroplastomic, containing both wild-type and altered plastid chromosomes. The homoplastomic state, in which all the wild-type copies are replaced by mutant chromosomes, is usually achieved after a few cycles of regeneration, during which both wild-type and mutant plastid chromosomes and the corresponding plastid types segregate. If the affected gene fulfils a vital task, the plant remains heteroplastomic, with wild-type and transformed plastid chromosomes co-existing at a relatively constant ratio under conditions selective for transgene expression (Drescher et al. 2000). Thus, to judge whether a plastid gene is dispensable or not, it is essential to prove that transplastomic knock-out mutants reach the homoplastomic state for the null allele. The complete absence of wild-type chromosome copies is also an indispensable requirement for obtaining stable and clearly interpretable phenotypes (e.g. due to a disrupted dispensable or modified gene). In contrast to unicellular algae, like C. reinhardtii (Rochaix 1995, 1997), that contain only a single plastid with a relatively small number of approximately 80 chromosome copies per cell, for the highly reiterated higher plant plastome with its 5,000–10,000 chromosomes per cell this task is far from trivial. Analysis is further complicated by the finding that higher plants contain promiscuous plastid DNA segments in both nucleus and mitochondria (Ellis 1982; Stern and Lonsdale 1982; Timmis and Scott 1983; Ayliffe and Timmis 1992; Unseld et al. 1997; Ayliffe et al. 1998; The Arabidopsis Genome Initiative 2000). These may appear analytically as residual wild-type plastid DNA copies, even when all non-transformed plastid chromosome copies have been segregated and eliminated. It is therefore not surprising that the issue of the homo-/heteroplastomic state of transplastomic mutants is a matter of controversy and essentially remains to be settled (Burrows et al. 1998; Kofer et al. 1998; Koop et al. 1998; Maliga and Nixon 1998; DeSantis-Maciossek et al. 1999; Mäenpää et al. 2000; Ruf et al. 2000; Baena-Gonzalez et al. 2001; Swiatek et al. 2001).
Two strategies are generally applied to monitor the presence of residual wild-type plastid chromosome copies in transplastomic plants: (1) Southern (RFLP) analysis of restriction fragment patterns differing between wild-type and mutated plastid chromosome copies and (2) PCR analysis, using primers flanking the insertion site. For fertile transplastomic plants, homoplastomy can alternatively be examined indirectly by analysing seeds in large-scale germination assays (Svab and Maliga 1993; Maliga and Nixon 1998).
In this communication, we describe the comparison of customary Southern and PCR approaches using DNA of total cells or gradient-purified plastid fractions versus a new strategy which is based on PCR analyses of pulsed-field gel electrophoresis (PFGE)-purified plastid chromosomes to evaluate the hetero- or homoplastomic state of transplastomic tobacco plants. Transformants disrupted in either ycf1, ycf9 or ycf10 were grown for repeated regeneration cycles to allow segregation of plastid genomes and plastid types and were analysed for the presence of residual wild-type plastid chromosome copies.
Of the basically three approaches used, only PCR analysis of plastid DNA purified by PFGE proved suitable to reliably judge the hetero-/homoplastomic state of transplastomic plants.
Materials and methods
Plant material
For plant transformation and molecular analyses, tobacco plants (Nicotiana tabacum L. cv Petit Havana) were axenically grown at 24 °C with 8 h/16 h dark/light cycles (using 100 µE m−2 s−1 standard light; Osram L85 W/25 universal white fluorescent lamps) in jars with agar-solidified B5 medium (Gamborg et al. 1968) supplemented with vitamins (per litre: 100 mg myo-inositol, 10 mg thiamine HCl, 1 mg pyridoxine HCl, 1 mg nicotinic acid) and 0.2% sucrose.
Construction of a recombinant plasmid for ycf10 inactivation
A 3,235-bp fragment of tobacco plastid DNA containing ycf10 was excised from plasmid pTB22 (Shinozaki et al. 1986) with KpnI (nucleotide position 62,075) and SalI (nucleotide position 65,310), ligated into vector pGEM-3Zf(−) (Promega, USA) and cloned in Escherichia coli DH5α. The ycf10 locus was inactivated by digestion with BbsI at nucleotide position 63,571 and insertion of the blunt-ended aadA cassette (Koop et al. 1996). Plasmid DNA from an individual bacterial colony carrying the aadA cassette in the same polarity as ycf10 was used for transformation of tobacco plastids.
Plastid transformation
The ycf10 construct containing the aadA cassette was introduced into tobacco plastid DNA by particle bombardment of tobacco leaves (Svab and Maliga 1993). Plastid transformation followed essentially the procedures described by DeSantis-Maciossek et al. (1999). Three independent mutant lines were selected for analysis and subjected to 3–20 regeneration cycles.
Southern analysis
Leaf DNA was isolated according to Doyle and Doyle (1990). For Southern analyses, 5 µg of total DNA from leaf material or gradient-purified plastids were restricted with EcoRI and the fragments electrophoretically separated in agarose gels. DNA transfer from the gel onto Nylon membranes (Hybond-N+; Amersham Pharmacia Biotech, Germany) was performed as described by Sambrook et al. (1989). Probes were radiolabelled (32P) with the Random primed DNA labelling kit (Roche Diagnostics, Switzerland), according to the manufacturer's protocol.
Isolation of plastids
Leaf material was homogenised in extraction buffer [0.33 M sorbitol, 25 mM Hepes, 25 mM 2-(N-morpholino)ethanesulfonic acid, 4 mM Na-ascorbate, 1.2 mM MnCl2, 0.8 mM MgCl2, 4 mM EDTA, 1 mM KH2PO4, 4 mM dithiothreitol, 0.2% (w/v) bovine serum albumin, 0.1% (w/v) polyvinylpyridine (PVP-10), pH 6.8]. Homogenates were filtered through Miracloth (Calbiochem, USA) and the filtrate was centrifuged at 4,000 g for 2 min. The pellet was resuspended in a buffer containing 0.33 M sorbitol and 50 mM Hepes/KOH (pH 7.6) and fractionated by discontinuous (30%+80% for mutant material, 40%+80% for the wild type) Percoll gradient centrifugation at 15,700 g for 20 min at 4 °C. Intact chloroplasts banding at the cushion interface were recovered and the plastids were washed and finally resuspended in 0.33 M sorbitol, 50 mM Hepes/KOH (pH 7.6).
Preparation of plastid lysates for PFGE
For PFGE, fractions containing intact chloroplasts, prepared as described above, were adjusted to a chlorophyll concentration of 5 mg ml−1 with isotonic medium and incubated for 5 min at 42 °C. Low-melting-point agarose (0.9%, for insert moulds; FMC, USA) in LMP buffer (125 mM EDTA, 330 mM sorbitol, 25 mM Na-citrate/HCl, pH 7.0, 90 mM β-mercaptoethanol) was dissolved at 80 °C and cooled to 42 °C before it was carefully mixed with the chloroplast suspension (300 µl of agarose per 100 µl of chloroplast suspension, containing approximately 5×108 chloroplasts, as revealed by microscopy). The chloroplast/agarose sol was loaded into insert moulds and stored for 1 h at 4 °C in the dark. Solidified blocks were washed for 15 min at room temperature in 1.5 ml (per block) buffer A (450 mM EDTA, pH 8.0, 10 mM Tris-HCl, pH 8.0, 1% sodium lauroyl sarcosinate) and incubated overnight in 1.5 ml buffer A containing 20 µg RNase A ml−1 at 37 °C, with gentle agitation. They were then incubated in 1.5 ml buffer A containing 0.2 mg Proteinase K ml−1 for 24 h at 50 °C, again with gentle agitation. After 24 h, the protease solution was removed and blocks were washed twice in 1.5 ml buffer B (500 mM EDTA, pH 9.0) for 4 h each time and then overnight at 50 °C. The blocks were finally transferred to 1.5 ml buffer B and stored at 4 °C until use.
Pulsed-field gel electrophoresis
The blocks with immobilised chloroplast remnants were loaded onto a 1% SeaKem LE agarose gel in 1× Tris-borate EDTA (TBE) buffer. Concatemers of λ-DNA (Amersham Pharmacia Biotech, Germany) were used as molecular weight markers. Gels were run at 170 V at 12 °C, using the hexagonal electrode array in a Pulsaphor device (model LKB 2015, Pharmacia, Germany; cf. Kleine et al. 1993). The pulse time gradient used started with 60 s and decreased to 0.1 s over a period of 24 h. The gel was then stained for 1 h in 150 ml TBE buffer with 7.5 µl ethidium bromide (10 mg ml−1), destained in water and photographed under UV light.
Isolation of DNA from the pulsed-field gel
The bands of monomeric and dimeric plastid DNA were excised from the gel with a clean scalpel blade. DNA was eluted from the agarose slice with the QIAEX II kit (Qiagen, Hilden, Germany), according to the manufacturer's protocol.
PCR amplification of plastid, mitochondrial and nuclear sequences
PCR was conducted by denaturing the DNA at 94 °C for 30 s, annealing the primers at 50 °C for 1 min and DNA synthesis at 72 °C for 2 min, for 31 cycles; and the products were analysed in 0.8% agarose gels. The following primers were used: 5′-GTGACATTTCCGTTCTTATG-3′ (ycf1a) and 5′-AGTCTGGATACGGCAAAATC-3′ (ycf1b) for ycf1, 5′-CTCAGTTAATAAATGTCTGGAA-3′ (ycf10forPF) and 5′-TGTATCTTGATTAATTGGAT-3′ (ycf10revPF) for ycf10, 5′-CATATAGAATCAATGGATTCAT-3′ (ycf9forPF) and 5′-ACAACGGGTACGCTAATCAA-3′ (ycf9revPF) for ycf9, 5′-CAATGGACGAGGTAGTCGTA-3′ (coxfor) and 5′-CTATTATAGTCCGAATACTCAT-3′ (coxrev) for mitochondrial DNA, 5′-CCCTCCTATTTGATGCGCACAC-3′ (Hex3P) and 5′-GGATGCATAGGGTATGCACGTCC-3′ (Hex9 M) for nuclear DNA, or 5′-TGCTGGCCGTACATTTGTACG-3′ (aadALI59) and 5′-CACTACATTTCGCTCATCGCC-3′ (aadARE60) for aadA.
Results
Targeted inactivation of tobacco plastid ycf1, ycf9 and ycf10 reading frames
Transplastomic ycf1, ycf9 and ycf10 mutants were obtained by targeted disruption of the respective reading frames in the tobacco plastid genome. The generation and functional analysis of the ycf1 and ycf9 mutants were described by Drescher et al. (2000) and Swiatek et al. (2001). To obtain ycf10-deficient mutants, a knock-out allele for tobacco ycf10 was constructed (Fig. 1). The recombinant DNA fragment, modified by integration of the spectinomycin resistence-conferring aadA cassette (Goldschmidt-Clermont 1991; Svab and Maliga 1993; Koop et al. 1996), was inserted at its target site within a background of tobacco plastid DNA appropriate for homologous recombination. The final construct was introduced into the plastid chromosome biolistically by bombarding sterile leaves with plasmid DNA-coated gold particles. Regenerated plants of three independently selected primary transformants resistant to spectinomycin were screened for correct insertion by PCR, Southern and nucleotide sequence analyses. To speed up segregation of wild-type and mutant plastid chromosomes, the lines were subcultured at early stages (2–3 weeks after shoot regeneration) and grown for up to 20 regeneration cycles on selective medium.

Construction of a plastid transformation vector for targeted inactivation of ycf10. The KpnI/SalI fragment of plasmid pTB22 (Shinozaki et al. 1986), containing ycf10 and its approximately 1-kb flanking regions, was cloned in vector pGEM-3Zf(−). Inactivation of the locus was performed by insertion of the aadA cassette at the gene-internal BbsI site. A plasmid with the aadA cassette inserted in the same orientation as ycf10 was used for plastid transformation. The probe used for Southern analysis is indicated below ycf10 as a thick black line. Small arrows correspond to the primer pairs employed for PCR analysis. Detailed descriptions of constructs for the inactivation of ycf9 (pΔycf9) and ycf1 (pΔycf1) are given by Swiatek et al. (2001) and Drescher et al. (2000), respectively
Judging the hetero-/homoplastomic state of transplastomic ycf1, ycf9 and ycf10 mutants
Southern and PCR analysis of gradient-purified plastids
DNA from gradient-purified chloroplast fractions was first checked for residual wild-type chloroplast DNA copies in transformed plants by Southern-type RFLP analyses. Whereas for Δycf9 material no wild-type signal was detectable after three cycles of shoot regeneration (data not shown; Swiatek et al. 2001), for ycf10 a faint signal corresponding to the wild-type restriction fragment of 580 bp was still detectable and did not disappear for seven further regeneration cycles (Fig. 2A). Due to its proposed indispensable function (Drescher et al. 2000), in Δycf1 material wild-type and mutant restriction fragments persisted at a relatively stable ratio even after more than ten regeneration cycles (data not shown). At present, the absence of wild-type RFLP signals in DNA from gradient-purified plastids as observed for Δycf10 and Δycf9 material would be a generally accepted criterion for homoplastomy of transplastomic mutants (e.g. Maliga and Nixon 1998; Ruf et al. 2000).

Analysis of DNA. A Southern analysis of DNA from wild-type and Δycf10 plants. DNA (5 µg) was restricted with EcoRI and the fragments were separated electrophoretically, blotted and hybridised with a radiolabelled ycf10 probe. Restriction of total wild-type DNA resulted in a 580-bp fragment, restriction of transformed DNA giving a 1.37-kbp fragment. Lane 1 Wild type (WT), lane 2 heteroplastomic mutant after three regeneration cycles, lane 3 virtually homoplastomic mutant material after ten regeneration cycles. B PCR analysis of total DNA of wild-type and Δycf10 mutant material (after ten regeneration cycles, as used in A, lane 3), using ycf10-specific primers. Lane M Molecular mass marker, lane 1 wild-type DNA, lane 2 Δycf10 DNA after ten regeneration cycles. Two products are visible, at 221 bp and 1.01 kbp (corresponding to wild-type and Δycf10 DNA, respectively)
However, application of the very same Δycf10 and Δycf9 DNA fractions to PCR analysis (the second approach, which is substantially more sensitive) led to amplification products characteristic for wild-type plastid chromosomes even after 10–20 regeneration cycles (Figs. 2B, 3A, B). These signals could either result from residual wild-type plastid chromosome copies in the transplastomic material and/or (at least in part) trace back to promiscuous DNA of extraplastidic origin. The decreased signal intensities for the wild-type amplification products with DNA from gradient-purified plastids, compared with total leaf DNA as a template, as found for ycf10 (Fig. 3A, lanes 5, 6) and ycf9 (Fig. 3B, lanes 2, 3), suggested, but did not prove, a non-plastid, promiscuous origin.

PCR analysis of plastid DNA. A PCR analysis of plastid DNA from wild-type and Δycf10 tobacco. Assays were performed using ycf10-specific primers with different DNA preparations as templates: T total DNA, GP DNA isolated from gradient-purified plastid fractions, PF plastid DNA isolated by pulsed-field gel electrophoresis. B PCR analysis of plastid DNA from Δycf9 tobacco, using ycf9-specific primers and the DNA fractions T, GP and PF, as described in A. C PCR analysis of plastid DNA from wild-type and Δycf1 tobacco. Since a large segment of the ycf1 reading frame was deleted for the construction of the mutant allele (Drescher et al. 2000), ycf1-specific primers were used for detection of the wild type and aadA-specific primers were used for detection of transformed chromosome copies in plastid DNA purified by pulsed-field gel electrophoresis. D PCR analysis to check for the presence of mitochondrial DNA in different DNA preparations of tobacco wild-type and Δycf10 mutant material. A coxII-specific primer pair was used to amplify DNA fragments originating in the mitochondrial genome, using T, GP and PF fractions, as described in A. E Analysis for the presence of nuclear DNA in different DNA preparations of tobacco wild-type and Δycf10 mutant material. A rpoH/I-specific primer pair was used in PCRs to amplify DNA fragments originating in the nuclear genome, using T, GP and PF fractions, as described in A. Amplification products were separated electrophoretically and visualised by ethidium bromide staining (A, B, D, E, left panels, C) or hybridisation to radiolabelled ycf10-, ycf9-, coxII- and rpoH/I-specific probes (A, B, D, E right panels, respectively). The molecular mass of amplification products is indicated at the right. Lane M Molecular mass marker, lane 1 no-template control
To evaluate the DNA fractions from transplastomic material, generally utilised to check for residual wild-type plastid chromosome copies or contamination with nuclear and/or mitochondrial DNA (the sources of promiscuous DNA), the approach was extended by employing compartment-specific primer pairs for coxII (encoding subunit II of mitochondrial cytochrome oxidase; Fig. 3D) and for the nuclear rpoH/I loci (encoding chloroplast-targeted phage-type RNA polymerases; Hedtke et al. 2002; Fig. 3E) in PCR assays. To enhance the detection limit of the amplified gene fragments and to allow quantitative comparisons, PCR products were separated by agarose gel electrophoresis, blotted onto Nylon membranes and probed with radioactively labelled, gene-specific probes.
With the primer pairs, diagnostic for mitochondrial or nuclear genes, the fraction of gradient-purified intact plastids clearly turned out to be contaminated by low amounts of both mitochondrial (Fig. 3D, lanes 3, 6) and nuclear DNA (Fig. 3E, lanes 3, 6), consistent with previous subcellular fractionation work (Herrmann 1982). From this, it is obvious that PCR, although highly sensitive, inherently cannot discriminate between promiscuous DNA and/or minute amounts of residual wild-type chromosomal copies and that a different strategy is essential to discern between these alternatives.
PCR analysis with plastid chromosomes fractionated by PFGE
In order to assess whether the wild-type amplification signals detected in Δycf10 and Δycf9 transplastomic material originated from incompletely segregated wild-type plastid chromosomes or DNA promiscuity, plastid chromosome fractions derived from PFGE were analysed. For PFGE analyses, gradient-purified intact chloroplasts were embedded in agarose blocks and processed by treatment with detergent and protease as described in Materials and methods. The immobilised DNA, including intact plastid chromosomes, was then size-fractionated in agarose gels with pulsed electrical fields into their monomeric and multimeric forms (Fig. 4; cf. Deng et al. 1989; Backert et al. 1995). DNA was subsequently eluted from agarose slices containing the plastid chromosome by silica particles (Qiagen) and analysed by PCR with primer pairs for ycf10, ycf9 and ycf1 (Fig. 3A, B C), for mitochondrial coxII (Fig. 3D) and for nuclear rpoH/I (Fig. 3E). In striking contrast to DNA isolated from gradient-purified plastid fractions (Figs. 3D, E, lanes 3, 6), neither mitochondrial products (Fig. 3D, lanes 4, 7) nor gene fragments of nuclear origin (Fig. 3E, lanes 4, 7) could be amplified from PFGE-purified plastid DNA. This substantiates the high purity of the preparations, which apparently lack even traces of contaminating mitochondrial and/or nuclear DNA.

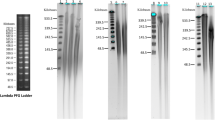
Pulsed-field gel electrophoresis of wild-type and Δycf10 tobacco plastid chromosomes. Gradient-purified intact plastids of wild-type and Δycf10 material were immobilised in low-melting point agarose blocks and the DNA separated in a pulsed electrical field. Different conformations of plastid DNA are visible (from bottom to top): monomer (156 kb), dimer (312 kb) and molecules of even higher molecular mass accumulating at the compression zone of the gel
When PFGE-fractionated plastid chromosomes from transplastomic plants served as a template for amplification, for both ycf10 and ycf9 mutants, no wild-type-specific PCR product could be detected (Fig. 3A, lane 7, B, lane 4). In contrast, wild-type signals could still be verified in relatively high amounts in ycf1 mutant material (Fig. 3C, lane 3). These results not only confirm the obligatory heteroplastomic state of ycf1 mutant plants (Drescher et al. 2000), they prove homoplastomy of Δycf10 and Δycf9 material, implying the dispensable function of the corresponding polypeptides under conditions when photosynthesis is non-essential. They also verify that wild-type signals derived from DNA of total leaf material and gradient-purified plastid fractions of ycf10 and ycf9 mutant plants do not originate in plastid chromosomes, but with high probability from promiscuous DNA in nuclei and/or mitochondria.
Estimation of template abundance
Promiscuous DNA of plastid origin is frequent (cf. Herrmann 1997) and has been detected both in the nucleus and in mitochondria in various plant species (Ellis 1982; Stern and Lonsdale 1982; Timmis and Scott 1983; Ayliffe and Timmis 1992; Unseld et al. 1997; Ayliffe et al. 1998; The Arabidopsis Genome Initiative 2000). To estimate the abundance of wild-type ycf10 template in the mutant material, PCR analysis was carried out with mixtures of varying ratios of plasmids containing either wild-type (pWt) or disrupted ycf10 (pMu). Such template mixtures would also be present in both heteroplastomic and homoplastomic tissues in the presence of promiscuous DNA. The PCRs, carried out with the same primers as those applied for mutant analysis, were designed to determine the lowest ratio of wild-type to mutant ycf10 that would still allow detection of an amplified wild-type PCR product. The ratios ranged from equimolar (5×107 copies each) to 1/106 molar with a constant quantity of 108 DNA templates per reaction. A corresponding number of molecules was estimated to have served as template in PCRs with PFGE-fractionated plastid chromosomes.
Co-amplification of the 221-bp wild-type ycf10 sequence along with the 1.01-kbp mutant ycf10 sequence was observed at molar ratios of 1/105 and above (Fig. 5). It is important to note that, due to the bias towards shorter amplification products in PCRs, only weak mutant signals were detected at pWt/pMu ratios above 1/102.

PCR analysis of plasmid DNA containing ycf10 (pWt) or ycf10aadA (pMu) gene insertions. Lane M Molecular mass marker, lane 1 pWt, lane 2 pMu, lane 3 pWt/pMu (molar ratio 1/1), lane 4 pWt/pMu (1/10), lane 5 pWt/pMu (1/102), lane 6 pWt/pMu (1/103), lane 7 pWt/pMu (1/104), lane 8 pWt/pMu (1/105), lane 9 pWt/pMu (1/106). Amplification products were separated electrophoretically and visualised by ethidium bromide staining (upper panel) or by hybridisation to a radiolabelled ycf10-specific probe (lower panel). The molecular mass of amplification products is indicated at the right
Following these results, the lack of wild-type PCR product with PFGE-purified plastid DNA but not with that of gradient-purified plastids points to an abundance of less than one wild-type template per 105 mutant templates in the former instance. Clearly, more than one wild-type-like template per 105 mutant templates on average is present in gradient-purified plastids (compare Fig. 3A, lane 6 with Fig. 5, lane 8). Assuming residual wild-type ycf10 templates to be of plastid origin and assuming a total plastid DNA copy number of approximately 104 per cell (cf. Herrmann and Possingham 1980; Bendich 1987), the abundance of wild-type ycf10 would be less than one copy per ten cells, implying that those tissues are homoplastomic. Moreover, assuming that the wild-type-like templates found with high abundance in total cellular DNA and in DNA from gradient-purified plastids are all of non-plastid origin, they are not likely to derive only from a single copy of this promiscuous DNA per haploid genome in the nucleus. Rather, they represent multiple copies in the nucleus and/or mitochondria.
Discussion
The genetic transformation of plastids of higher plants serves as a powerful approach for basic research and biotechnological applications. The homoplastomy of transformants, i.e. the complete replacement of wild-type plastid chromosomes by transformed copies, is usually achieved by segregation of plastomes during repeated regeneration cycles under conditions selective for the modified plastid genome.
The presence of residual wild-type chromosome copies in transplastomic plants is generally tested by Southern and PCR analysis with total cellular DNA or DNA from gradient-purified plastids. Southern analysis, however, cannot reliably prove the absence of a certain plastid chromosome-type in primary heteroplastomic plant material (Moll et al. 1990). Although the PCR approach is superior in sensitivity to RFLP analysis and allows the detection of only minute numbers of wild-type DNA copies unverifiable by Southern experiments, it bears the risk of (co)amplifying promiscuous plastid DNA sequences residing in nucleus and/or mitochondria. This risk can be reduced, but not eliminated, by using DNA extracted from plastids that were purified by isopycnic, isotonic density gradient centrifugation as a template for amplification, since some nuclear chromatin and often mitochondria contaminate plastids, even after gradient centrifugation (Fig. 3D, E, lanes 2, 3, 5, 6; Herrmann 1982), mimicking genuine plastid chromosomes in PCR analyses. This precludes a clear statement whether a transplastomic material is free of residual wild-type plastid chromosome copies or not.
In this work, we could show that PFGE-purified plastid DNA serves as an ideal source to reliably judge the homoplastomic state of transplastomic plants. Size fractionation of plastid chromosomes (monomeric and multimeric conformations, Fig. 4) by PFGE and subsequent elution of the plastid chromosome fractions from the gel clearly led to the separation from contaminating DNA molecules of nuclear and mitochondrial origin. PCR analyses with primers specific for plastid, mitochondrial and nuclear genes pointed out the high purity of the plastid DNA fraction derived (Fig. 3) and allowed the detection of less than one wild-type plastid chromosome per ten transplastomic cells (Fig. 5). It should be noted that minor fractions of rearranged aberrant transgenic plastid chromosomes of anomalous sizes, as described by Svab and Maliga (1993), were not traced by this strategy. In cases where seed can be raised, an alternative strategy (which is technically easier and highly sensitive but more time-consuming) to verify the homoplastomy of transplastomic mutants is to test for retention of the selectable marker by germinating the seed on antibiotic-containing medium (Maliga and Nixon 1998). Only when homoplastomic for the transformed plastid chromosome, would all seeds be insensitive to antibiotic treatment.
At present, the PFGE-based approach was performed with eight transplastomic materials (disrupted in clpP, psbE, psbF, psbL, psbJ, ycf1, ycf9, ycf10), of which three different knock-out mutants affected in the genes ycf1, ycf9 or ycf10 (representatives of different segregation types: not segregating in the case of ycf1, slowly and quickly segregating in the case of ycf10 and ycf9, respectively) were chosen here as examples.
Δycf1 plants were reported to retain wild-type plastid chromosome copies when cultivated under heterotrophic in vitro culture conditions selective for transgene (aadA) expression, because of an essential but so far unknown function of the locus (Drescher et al. 2000). In this case, heteroplastomy of transformants was verified not only by RFLP analysis but also by test of seed progeny (Drescher et al. 2000). Our finding that the fraction of PFGE-purified plastid chromosomes of ycf1-mutants contains significant amounts of both wild-type and modified molecules (Fig. 3C) confirms this mutant is heteroplastomic and emphasises the role of ycf1 as an essential housekeeping gene.
Disruption of the ycf9 locus shows that its product specifies a constituent polypeptide of photosystem II (designated psbZ; Swiatek et al. 2001). Since photosynthesis is not required for tobacco grown heterotrophically on sucrose-containing tissue culture media, this mutant should be able to reach a homoplastomic state under those conditions. However, even after repeated regeneration cycles, PCR-based psbZ wild-type signals were identified, which suggested an (additional) indispensable function (Mäenpää et al. 2000; Baena-González et al. 2001). Analysis of PFGE-purified plastid chromosomes (Fig. 3B, lane 4), as performed in this work, clearly demonstrated the homoplastomic state of ycf9-deficient plants, which is in line with its functional role in photosynthesis. We could further show that wild-type ycf9 signals derived by analysis of DNA from total leaf material and even the gradient-purified plastid fraction (Fig. 3B, lanes 2, 3; cf. Mäenpää et al. 2000; Ruf et al. 2000; Baena-González et al. 2001) trace back to promiscuous ycf9 sequences, either in the nuclear or mitochondrial genome.
Finally, ycf10 represents a plastid gene, for which the function is still a matter of debate. The gene product, initially designated HBP (heme-binding protein; Willey and Gray 1990) was renamed CemA (chloroplast envelope membrane protein A) after its immunolocalisation in the inner chloroplast envelope membrane (Sasaki et al. 1993). The cyanobacterial cotA gene product, which displays high sequence identity to the Ycf10 polypeptide from higher plants, has been shown to mediate light-induced proton extrusion (Katoh et al. 1996a, 1996b). In contrast, a role in the inorganic carbon uptake into chloroplasts has been proposed, based on inactivation of ycf10 in plastids of C. reinhardtii (Rolland et al. 1997). As shown in this work, Δycf10 tobacco mutants are able to reach the homoplastomic state when grown on sucrose, which is in line with the proposed function of ycf10 in Chlamydomonas (Katoh et al. 1996a, 1996b; Rolland et al. 1997). Again, as shown for Δycf9, wild-type signals are detectable by PCR, using DNA from total leaf material or gradient-purified plastid fractions (Fig. 3A, lanes 5, 6). Quantitative analysis revealed that these wild-type ycf10-signals must originate from multiple copies of promiscuous DNA in the nucleus and/or mitochondria (Fig. 5).
The presence of promiscuous plastid DNA in the nuclear and mitochondrial genome generally aggravates verification of the homoplastomy of transplastomic mutants by PCR and Southern analysis, even with DNA from gradient-purified plastids, since these DNA preparations are generally contaminated with traces of nuclear and mitochondrial DNA (Fig. 3D, E). Our data illustrate that PFGE-purified plastid DNA provides a reliable means of discriminating between residual wild-type plastome copies and promiscuous DNA: wild-type templates identified by PCR with PFGE-purified plastid DNA clearly originate from plastid chromosomes and not from promiscuous DNA. Therefore, the lack of wild-type amplification products proves homoplastomy for mutant plastomes. Our approach thus overcomes a significant problem in the determination of homoplastomy in plastome transformation.
References
Ayliffe MA, Timmis JN (1992) Tobacco nuclear DNA contains long tracts of homology to chloroplast DNA. Theor Appl Genet 85:229–238
Ayliffe MA, Scott NS, Timmis JN (1998) Analysis of plastid DNA-like sequences within the nuclear genomes of higher plants. Mol Biol Evol 15:738–745
Backert S, Dörfel P, Börner T (1995) Investigation of plant organellar DNAs by pulsed-field gel electrophoresis. Curr Genet 28:390–399
Baena-Gonzalez E, Gray JC, Tyystjarvi E, Aro EM, Mäenpää P (2001) Abnormal regulation of photosynthetic electron transport in a chloroplast ycf9 inactivation mutant. J Biol Chem 276:20795–20802
Bendich AJ (1987) Why do chloroplasts and mitochondria contain so many copies of their genome? BioEssays 6:279–282
Bock R (2001) Transgenic plastids in basic research and plant biotechnology. J Mol Biol 312:425–438
Bogorad L (2000) Engineering chloroplasts: an alternative site for foreign genes, proteins, reactions and products. Trends Biotechnol 18:257–263
Boynton JE, Gillham NW, Harris EH, Hosler JP, Johnson AM, Jones AR, Randolph-Anderson BL, Robertson D, Klein TM, Shark KB, Sanford JC (1988) Chloroplast transformation in Chlamydomonas with high velocity microprojectiles. Science 240:1534–1538
Burrows PA, Sazanov LA, Svab Z, Maliga P, Nixon PJ (1998) Identification of a functional respiratory complex in chloroplasts through analysis of tobacco mutants containing disrupted plastid ndh genes. EMBO J 17:868–876
Daniell H, Khan MS, Allison L (2002) Milestones in chloroplast genetic engineering: an environmentally friendly era in biotechnology. Trends Plant Sci 7:84–91
Deng XW, Wing RA, Gruissem W (1989) The chloroplast genome exists in multimeric forms. Proc Natl Acad Sci USA 86:4156–4160
DeSantis-Maciossek G, Kofer W, Bock A, Schoch S, Maier RM, Wanner G, Rüdiger W, Koop HU, Herrmann RG (1999) Targeted disruption of the plastid polymerase genes rpoA, rpoB, rpoC1: molecular biology, biochemistry and ultrastructure. Plant J 18:477–489
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Drescher A, Ruf S, Calsa T Jr, Carrer H, Bock R (2000) The two largest chloroplast genome-encoded reading frames of higher plants are essential genes. Plant J 22:97–104
Ellis J (1982) Promiscuous DNA—chloroplast genes inside plant mitochondria. Nature 299:678–679
Gamborg OL, Miller RA, Ojima K (1968) Nutrient requirements of suspensions of soybean root cells. Exp Cell Res 50:151–158
Golds T, Maliga P, Koop HU (1993) Stable plastid transformation in PEG-treated protoplasts of Nicotiana tabacum. Biotechnology 11:95–97
Goldschmidt-Clermont M (1991) Transgenic expression of aminoglycoside adenyl transferase in the chloroplast: a selectable marker for site-directed transformation of Chlamydomonas. Nucleic Acids Res 19:4083–4089
Hedtke B, Legen J, Weihe A, Herrmann RG, Börner T (2002) Six active phage-type RNA polymerase genes in Nicotiana tabacum. Plant J 30:625–637
Herrmann RG (1982) The preparation of circular DNA from plastids. In: Edelman M, Hallick RB, Chua WH (eds) Methods in chloroplast molecular biology. Elsevier, Amsterdam, pp 259–280
Herrmann RG (1997) Eukaryotism, towards a new interpretation. In: Schenk HEA, Herrmann RG, Jeon KW, Müller NE, Schwemmler W (eds) Eukaryotism and symbiosis. Springer, Berlin Heidelberg New York, pp 73–118
Herrmann RG, Possingham JV (1980) Plastid DNA—the plastome. In: Reinert J (ed) Results and problems in cell differentiation, vol 10. Springer, Berlin Heidelberg New York, pp 45–96
Katoh A, Lee KS, Fukuzawa H, Ohyama K, Ogawa T (1996a) cemA homologue essential to CO2 transport in the cyanobacterium in Synechocystis PCC 6803. Proc Natl Acad Sci USA 96:4006–4010
Katoh A, Sonoda M, Katoh H, Ogawa T (1996b) Absence of light-induced proton extrusion in a cotA-less mutant of Synechocystis sp. strain PCC 6803. J Bacteriol 178:5452–5455
Khan MS, Maliga P (1999) Fluorescent antibiotic resistance marker for tracking plastid transformants in higher plants. Nat Biotechnol 17:910–915
Kindle KL, Richards KL, Stern DB (1991) Engineering the chloroplast genome: techniques and capabilities for chloroplast transformation in Chlamydomonas reinhardtii. Proc Natl Acad Sci USA 88:1721–1725
Kleine M, Michalek W, Graner A, Herrmann RG, Jung C (1993) Construction of a barley (Hordeum vulgare L.) YAC library and isolation of a Hor1-specific clone. Mol Gen Genet 240:265–272
Kofer W, Koop HU, Wanner G, Steinmüller K (1998) Mutagenesis of the genes encoding subunits A, C, H, I, J and K of the plastid NAD(P)H-plastochinone-oxidoreductase in tobacco by polyethylene glycol-mediated plastome transformation. Mol Gen Genet 258:166–173
Koop HU, Kofer W (1995) Plastid transformation by polyethylene glycol treatment of protoplasts and regeneration of transplastomic tobacco plants. In: Potrykus I, Spangenberg G (eds) Gene transfer to plants. Springer, Berlin Heidelberg New York, pp 75–82
Koop HU, Steinmüller K, Wagner H, Rossler C, Eibl C, Sacher L (1996) Integration of foreign sequences into the tobacco plastome via polyethylene glycol-mediated plastom transformation. Planta 199:193–201
Koop HU, Kofer W, Steinmüller K (1998) Judging the homoplastomic state of plastid transformants—reply. Trends Plant Sci 3:377–378
Mäenpää P, Gonzales EB, Chen L, Khan MS, Gray JC, Aro EM (2000) The ycf9 (orf62) gene in the plant chloroplast genome encodes a hydrophobic protein of stromal thylakoid membranes. J Exp Bot 51:375–382
Maliga P (2002) Engineering the plastid genome of higher plants. Curr Opin Plant Biol 5:164–172
Maliga P, Nixon PJ (1998) Judging the homoplastomic state of plastid transformants. Trends Plant Sci 3:376–377
Moll B, Polsby L, Maliga P (1990) Streptomycin and lyncomycin resistances are selective plastid markers in cultured Nicotiana cells. Mol Gen Genet 221:245–250
Rochaix JD (1995) Chlamydomonas reinhardtii as the photosynthetic yeast. Annu Rev Genet 29:209–230
Rochaix JD (1997) Chloroplast reverse genetics: new insights into the function of plastid genes. Trends Plant Sci 2:419–425
Rolland N, Dorne AJ, Amoroso G, Sültemeyer DF, Joyard J, Rochaix JD (1997) Disruption of the plastid ycf10 open reading frame affects uptake of inorganic carbon in the chloroplast of Chlamydomonas. EMBO J 16:6713–6726
Ruf S, Biehler K, Bock R (2000) A small chloroplast-encoded protein as a novel architectural component of the light-harvesting antenna. J Cell Biol 149:369–378
Ruf S, Hermann M, Berger IJ, Carrer H, Bock R (2001) Stable genetic transformation of tomato plastids and expression of a foreign protein in fruit. Nat Biotechnol 9:870–875
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Sasaki Y, Sekiguchi K, Nagano Y, Matsumo R (1993) Chloroplast envelope protein encoded by the chloroplast genome. FEBS Lett 316:93–98
Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, Zaita N, Chunwongse J, Obokata J, Yamaguchi-Shinozaki K, Ohto C, Torazawa K, Meng BY, Sugita M, Deno H, Kamogashira T, Yamada K, Kusuda J, Takaiwa F, Kato A, Tohdoh N, Shimada H, Sugiura M (1986). The complete nucleotide sequence of tobacco chloroplast genome: its gene organization and expression EMBO J 5:2043–2049
Sidkar SR, Serino G, Chaudhuri S, Maliga P (1998) Plastid transformation in Arabidopsis thaliana. Plant Cell Rep 18:20–24
Sidorov VA, Kasten D, Pang SZ, Haidukiewicz PTJ, Staub JM, Nehra NS (1999) Stable chloroplast transformation in potato: use of green fluorescent protein as a plastid marker. Plant J 19:209–216
Stern DB, Lonsdale DM (1982) Mitochondrial and chloroplast genomes of maize have a 12-kilobase DNA sequence in common. Nature 299:698–702
Svab Z, Maliga P (1993) High-frequency plastid transformation in tobacco by selection for a chimeric aadA gene. Proc Natl Acad Sci USA 90:913–917
Svab Z, Hajdukiewicz P, Maliga P (1990) Stable transformation of plastids in higher plants. Proc Natl Acad Sci USA 87:8526–8530
Swiatek M, Kuras R, Sokolenko A, Higgs D, Olive J, Cinque G, Muller B, Eichacker LA, Stern DB, Bassi R, Herrmann RG, Wollman FA (2001) The chloroplast gene ycf9 encodes a photosystem II (PSII) core subunit, PsbZ, that participates in PSII supramolecular architecture. Plant Cell 13:1347–1367
The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796–815
Timmis JN, Scott NS (1983) Spinach nuclear and chloroplast DNAs have homologous sequences. Nature 305:65–67
Unseld M, Marienfeld JR, Brandt P, Brennicke A (1997) The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat Genet 1:57–61
Willey DL, Gray LC (1990) An open reading frame encoding a putative haem-binding polypeptide is co-transcribed with the pea chloroplast gene for apocytochrome f. Plant Mol Biol 15:347–356
Acknowledgements.
We thank Ralph Bock for providing the transplastomic Δycf1 line and Christian Schmitz-Linneweber for critical reading of the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 184) and by the Fonds der Chemischen Industrie.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F.-A. Wollman
Rights and permissions
About this article
Cite this article
Swiatek, M., Greiner, S., Kemp, S. et al. PCR analysis of pulsed-field gel electrophoresis-purified plastid DNA, a sensitive tool to judge the hetero-/homoplastomic status of plastid transformants. Curr Genet 43, 45–53 (2003). https://doi.org/10.1007/s00294-003-0369-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-003-0369-4