
Abstract
The polymerization process of the dual-cured resin cement AllCem (FGM, Joinvile, Brazil) was investigated by unilateral nuclear magnetic resonance (NMR-MOUSE) and electron paramagnetic resonance (EPR). The NMR experiment allows measurements of the mobile monomers as function of time and depth, while real-time X-band EPR monitoring gives information about the concentration of free radicals. The monomer concentration instantly decays for photo-cured samples, but it remains constant about ~4 min for self-cured samples before starting to decrease. For the self-cured sample, the concentration of free radicals suddenly decreases to zero and then reappears with a strongly increasing rate. Models for the polymerization kinetics of each initiation protocol are proposed, giving insight into the polymerization process of the dual-cured resin cement.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A composite is generally defined as a material composed of two or more distinct phases. Dental composites consist of a polymerizable resin base containing a ceramic filler that does not interfere on the polymerization reaction. Most dental polymers are based on acrylic resins made up of monomethacrylate, dimethacrylate, or trimethacrylate monomers [1]. The most common restorative materials are resin composites based on the Bis-phenol-A-bis(glycidyl methacrylate) (Bis-GMA), which copolymerizes with triethylene glycol dimethacrylate (TEGDMA). Also, the dimethacrylate monomers Bis-phenol-A-dimethacrylate (Bis-EMA) and a urethane dimethacrylate (UDMA) are commonly used. These composite resins polymerize by the absorption of blue visible light which activates the photoinitiator camphorquinone (CQ) from the ground state to an excited state where it reacts with a hydrogen donor and produces an initiating free radical (FR). Thus, the excited molecule interacts with other molecules to form FRs capable of starting the radical polymerization by rapid addition of monomers to the chain [1].
Self-curing composites are two-component filled systems based on dimethacrylate monomers, such as Bis-GMA, TEGDMA, and/or UDMA with an organic peroxide, usually benzoyl peroxide (BPO). To promote the decomposition of BPO at oral temperature, cold-cure acrylic resins need another chemical, usually an aromatic tertiary amine to carry out the redox initiation, together with BPO in a short period of time [2, 3]. Those amines are the co-initiator. Amines are generally skin-sensitive, so medical and clinical tests are made by manufacturers to ensure safety for patients and users. Despite the differences in appearance, the chemical initiation system of self-cure composites is virtually the same as self-cure acrylic resins (i.e., BPO/amine redox systems) [1].
Besides composite resins used to restore the lost part of the tooth, acrylic resin cements are used to retain intraradicular post for the purpose of assisting the retention of the restoration material. In general, dental resin cements are dual-cured to ensure polymerization at deepest points of the restoration where radiation cannot excite CQ, such that photo-cure and self-cure happen simultaneously and independently [4–8]. Because of clinical implications, an inhibitor is also added in the composition to guarantee that the material will not polymerize during storage and to provide the necessary working time to the professional.
The degree of conversion of monomers into polymer is related to the amount of FRs generated during the initiation step of the polymerization reaction, either by irradiation or benzoyl peroxide degradation. Incomplete polymerization compromises the mechanical properties of the composite material and final result of restoration. Understanding the polymerization process is important and can help the development of new materials. Since the FRs generated are paramagnetic species, electron paramagnetic resonance (EPR) spectroscopy is a reliable technique for identifying and quantifying the concentration of FRs in the sample without any changes in the materials′ composition [4, 5, 9]. The NMR-MOUSE (Nuclear Magnetic Resonance-Mobile Universal Surface Explorer) is a stray-field NMR device suitable to detect the monomer concentration and changes in molecular mobility non-destructively in real time [10, 11]. The NMR-MOUSE can be used to acquire depth profiles locally into the object by varying the distance between the NMR-MOUSE and the object [10, 11].
The purpose of this research is associating nuclear to electron magnetic resonance to probe the dual-cure of a commercial dental resin cement. The photo-curing and self-curing reactions were studied with the NMR-MOUSE technique, and the concentration of FRs was investigated by X-band electron paramagnetic resonance (EPR) spectroscopy. The time- and depth-dependent NMR signals of the photo-cure were modeled with the polymerization kinetics of the reactions. An empirical equation for describing the time-dependent monomer concentration is obtained for the self-cure reaction.
Experimental
Materials
Samples of dual-cured resin cement from Allcem (FGM, Joinville, Brazil) in the color shade A1 were examined with the NMR-MOUSE and X-band EPR spectroscopy. The initial compounds are two pastes separated in a dual-body syringe which are mixed in a self-mixing nozzle that accompanies the kit. The polymerization reaction begins upon mixing the base paste with the catalyst in the nozzle, and it can be accelerated by blue light irradiation. In all experiments, the initial paste and catalyst were mixed in a 1:1 weight ratio according to manufacturer’s recommendation. The particular resin cement was chosen for this study because it has the same composition and showed similar properties and quality compared to others used worldwide for the purpose of translucent fiberglass post cementation. The irradiation of the cement with the blue visible light was produced by a LED (Ultra Blue, Dabi Atlante, Ribeirão Preto, Brazil) with a potency of 492 mW/cm2. The resin cement samples were separated in two major groups: irradiated for 40 s and non-irradiated.
According to manufacturer’s information, the composition of the resin cement is Bis-phenol-A-bis(glycidyl methacrylate) (Bis-GMA), triethylene glycol dimethacrylate (TEGDMA), and Bis-phenol-A-dimethacrylate (Bis-EMA), initiators, coiniciators (camphorquinone and dibenzoil peroxide) and stabilizers. Barium-aluminosilicate glass microparticles and silicon dioxide nanoparticles are employed as fillers and inorganic charge. However, as the resin cement analyzed in this paper is a commercial material, information on the concentration of each chemical is not available.
Experiments
Unilateral nuclear magnetic resonance (NMR-MOUSE)
Using the proper self-mixing nozzle of the AllCem dual-body syringe, the resin cement samples were injected in a 7.6-mm-high cylindrical glass container with a 4.4 mm inner diameter. All NMR measurements were carried out with the stray-field sensor NMR-MOUSE PM 5 (Magritek GmbH, Aachen, Germany), which collects NMR signal from a slice 5 mm above the sensor surface. Coupled with a surface coil, which generates a radio-frequency magnetic field perpendicular to the static magnetic field, the 1H frequency of the NMR-MOUSE was 18.1 MHz with gradient 22 T/m at 5 mm [11]. The NMR-MOUSE sensor was operated by a Bruker Minispec spectrometer operating at the Larmor frequency. The 90° pulse length was set to 8 µs, the echo time was 32 µs, the acquisition window of the echo was 6 µs, and the recycle delay was adjusted to 0.15 s. Following previous work [10], 18 common glass slides with a thickness of 1 mm were used as light attenuator to slow down the curing time in order to follow the curing reaction in real time. As the number of scans was fixed to 128, a T 2 measurement took 25 s. The continuous T 2 measurements are an average of the signal changing during 25 s. This approximation is acceptable when changes are weak, for example, during the stationary state of a photo-curing reaction [10]. The space scale is always the distance from the sample surface closest to the sensor, set to 6.1, 6.6, 7.1, and 7.6 mm (Fig. 1). The sensitive volume is the region where the magnetic field lines are more uniform and allows acquiring NMR signal. The NMR data were acquired with the Carr–Purcell–Meiboom–Gill (CPMG) multi-echo trains [11]. The NMR echoes arise only from the mobile monomers present in the sample, while the signal from the solid polymer rapidly relaxes within about 32 µs during the dead time of the sensor [10]. All experiments were carried out at room temperature.

Schematic drawing of the NMR-MOUSE experiments
Electron paramagnetic resonance (EPR)
The concentration of FRs was determined by X-band EPR spectroscopy in real time during the reaction and correlated with the decay curve of monomers. As the EPR signal intensity is proportional to the amount of paramagnetic species in the sample, the second integral of the spectrum gives the number of FRs at the time of measurement. This is a reliable method for determining the amount of radical species in any sample. The EPR experiment does not give information about layers and positions in the sample as does the NMR-MOUSE. The EPR spectrum is always correspondent to the overall mass of the sample inside the resonant cavity. The X-band (~9 GHz) EPR spectra were obtained with a JEOL JES-PE-3X spectrometer at room temperature using 1 mW microwave power, 0.40 mT modulation amplitude, and 100 kHZ modulation frequency to avoid signal saturation. A JEOL standard sample of MgO:Mn2+ was used as intensity standard and g marker. The samples were placed in a 2 × 2 mm Teflon mold. The data treatment was performed with OriginLab software.
Polymerization reaction of the dual-cured resin cement
Polymerization mechanism of the self-cure
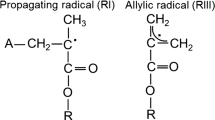
In the self-cured process, the BPO is degraded releasing two primary radicals and an inert product [2, 12]. Figure 2 represents the BPO degradation by interaction with the tertiary amine 4-(N,N-dimethylamino) phenethyl alcohol (DMPOH) [2, 12]. The primary FRs generated in the self-cure are the N-methylene radical (named PRI) and the benzoyloxy radical (named PRII), while the inert product is benzoic acid [2, 12]. Subsequently, PRI and PRII react with monomers leading to the induction step and then to the propagation step, where the active monomer reacts with other monomer molecules. The PRI reacts with monomers by breaking the double bonding or by hydrogen abstraction (Fig. 3a, b). The reaction generates two different radicals, the propagating radical (RI) and the allylic radical (RII), which can be detected by EPR spectroscopy [13]. Figure 4a, b represents the reaction of the PRII with methacrylate monomer generating two different radicals, RIII and RIV.

The degradation process of BPO generating two primary radicals, PRI (N-methylene radical) and PRII (benzoyloxy radical), and one inert product

a The reaction of PRI with a MMA monomer generating the propagating radical (RI). b Reaction of PRI with a MMA monomer by hydrogen abstraction generating an inert product and an allylic radical (RII)

a The reaction of PRII with a MMA monomer generating the propagating radical (RIII). b Reaction of PRII with a MMA monomer by hydrogen abstraction generating an inert product and an allylic radical (RIV)
The two different primary radicals generated during the initiation step are assumed to be capable of reacting with a monomeric double bond and initiate polymerization. In a previous paper, it was confirmed by multifrequency (X-, Q-, and W-band) EPR spectroscopy and spectra simulations that there are two populations of “live” macroradicals in the dual-cured sample: those which are active and are responsible for the polymerization continuity (propagating radical), and those which are trapped in the polymer network (allylic radical) [13]. Live radicals terminate by combination/disproportionation, primary radical termination, and chain transfer to amine [12, 13]. A great concentration of inhibitors is included to prevent early polymerization and proportionate working time.
Considering the symbols I and A to denote the peroxide and amine initiator molecules, respectively, M the monomer unit, and \({\text{RM}}_{n}^{*}\) the active macroradical, the reaction mechanism is presented in Table 1 for the self-polymerization of multifunctional monomers [12].
In this kinetic scheme, the symbols \(A^{*}\) and \(I^{*}\) represent the radicals formed by fragmentation of amine and benzoyl peroxide, respectively; \({\text{RM}}_{{n,{\text{b}}}}^{*}\) is used to specify the trapped macroradical containing n monomer units; Z is the inhibitor; and finally \(D_{n}\) stands for the “dead” polymer formed containing n monomer units [12].
Based on the general kinetics mechanism presented for the self-cured resin cement, rate equations for all the reacting species can be written as follows [12] Initiators:
Primary initiator radicals
Double bonds
Active macroradicals
Trapped radicals
Inhibitor
In these equations, \([{\text{RM}}^{*} ] = \mathop \sum \nolimits_{n = 1}^{\infty } {\text{RM}}_{n}^{*}\) and k b is the radical trapping rate constant, which is assumed to be dependent on the fractional free volume of the mixture (V f) according to \(k_{\text{b}} = k_{\text{b0}} \frac{1}{{{\text{e}}^{{{\raise0.7ex\hbox{${ - \gamma_{\text{b}} }$} \!\mathord{\left/ {\vphantom {{ - \gamma_{\text{b}} } {V_{\text{f}} }}}\right.\kern-0pt} \!\lower0.7ex\hbox{${V_{\text{f}} }$}}}} }}\), where k b0 is the pre-exponential factor and γ b is the dimensionless activation volume which governs the rate at which radical trapping increases as a function of fractional free volume. This set of differential ordinary equations can be numerically integrated with the Gear’s method [14] to give the time dependence of all reacting species.
During the initial stage of polymerization, where the effect of diffusion-controlled phenomena on the reaction rates is negligible and the steady-state approximation for macroradicals holds, the rate of polymerization (\(R_{\text{p}}\)) is given by [12, 15]
where \(k_{\text{d}}\) is the initiator decomposition rate constant, \(k_{\text{p}}\) and \(k_{\text{t}}\) represent the propagation and termination rate constant, respectively, and \(f_{1}\) and \(f_{2}\) denote the efficiency factors.
Polymerization mechanism of the photo-cure
In addition to the self-cure, the dual-cured resin cement has the photo-cured initiators added to its composition. The photo-initiation mechanism by the CQ reaction with a tertiary amine is well described by Truffier-Boutry et al. [16] for the composite resin. As the EPR spectrum does not depend on the inorganic charges and the photo-curing takes place independently of the self-cure, the model proposed by Truffier-Boutry et al. [16] is applicable to this case, such that the FRs responsible for the continuity of the polymerization initiated by photo-curing are the propagating (CH3–C*–CH2) and the allylic (CH2–C*–CH2) radicals, as shown by Fontes et al. [9] (Fig. 5). In addition, Vicentin et al. [13] showed that the nine-line EPR signal is independent on the initiation mode for a dual-cured resin cement.

Second step of the initiation reaction and the two different radicals formed for the photo-cured resin cement
The initiator (I) is activated by light irradiation either to a singlet (life time in the nano-second range) or a triplet state (with a life time of 50 ms) [10, 17, 18]. Triplet-activated CQ molecules (\(I_{\text{triplet}}^{*}\)) may react with the accelerator molecules \((A)\) and form two radicals [9, 16], \(I^{*}\) and \(R^{*}\). \(I^{*}\) shows little reactivity and the polymerization rate is determined by accelerator radical \(R^{*}\) reacting with the first monomer M. In the propagation stage, the macromolecular chains are formed by successive reaction with monomers. The reaction is terminated typically by combination and/or disproportionation (Table 2) [10, 17, 18].
In these reactions, \(k_{\text{i}}\), \(k_{\text{p}}\) and \(k_{\text{t}}\) denote the reaction rate constant of the initiation, propagation, and termination steps, and \(D_{m + n}\) stands for the “dead” polymer formed containing \(m + n\) monomer units.
The following classical analytical equations are developed for photo-cured kinetics for linear-chain systems [17]. If \(\varphi\) denotes the extinction coefficient assuming an infinite reservoir of accelerator molecules, \(I_{0}\) the intensity of light absorbed in the surface, \(\alpha\) the initiation efficiency, and d the length of the light path in the sample, the rate of production of primary radicals from the photo-sensitizer (\(R_{\text{PR}}\)) may be expressed by [10, 18]
A detailed kinetic analysis of the activation steps has been made by Cook [19]. The rate of production of primary radicals can be re-expressed via the following equation
where \(\beta\) is the fraction of exciplex “molecules” forming FR. Assuming that the rates of production and consumption of initiator radicals rapidly become equal (steady-state assumption), then \(R_{\text{PR}}\) is equal to the rate of initiation \(R_{\text{i}}\), meaning \(R_{\text{PR}} = R_{\text{i}}\) [17]. Approximating the rate of the bimolecular propagation reaction to be independent of the chain length, the propagation rate \(R_{\text{p}}\) may be expressed by
where \([M]\) is the monomer concentration and \([{\text{RM}}^{*} ]\) the concentration of growing chains. The reaction rate for termination by combination and/or disproportionation is written as
In these reactions, \(k_{\text{i}}\), \(k_{\text{a}}\), \(k_{\text{p}}\) and \(k_{\text{t}}\) denote the reaction rate constant of the initiation, rate constant of formation of exciplex from the bimolecular reaction between amine and CQ, and reaction rate of the propagation and termination steps. Assuming a steady-state, initiation and termination reaction rates are identical (\(R_{\text{i}} = R_{\text{t}}\)). Then the steady-state propagation rate of radical polymerization is given by
This leads to the time-dependent monomer concentration, \([M](t)\),
where \(t_{0}\)is the initial time, \([M]_{0}\) the initial monomer concentration, and \(\tau_{\text{P}} = \left[ {k_{\text{p}} \left( {\frac{{k_{i} I_{0} \alpha e^{ - \varphi d} }}{{2k_{\text{t}} }}} \right)^{1/2} } \right]^{{ - \frac{2}{3}}}\) the characteristic time constant for the photo-cure [10].
Results
Figure 6 shows the sum of the 64 first spin echoes in the CPMG detection trains at different depths versus the curing time acquired with single-sided NMR for the photo-cured (Fig. 6a) and self-cured samples (Fig. 6b). Figure 7 shows the intensity of the X-band EPR spectra obtained during 10 min for the self-cured sample and Fig. 8 for the photo-cured sample.

Echo sums as function of time and depth corresponding to the distance to the sensor a for the photo-cured sample and b for the self-cured sample

a EPR spectra of the self-cured sample obtained every 10 s. b Intensity of the EPR spectra of the self-cured sample obtained every 10 s in a time interval of 10 min

Intensity of the EPR spectra of the photo-cured sample
Discussion
The self-cured reaction starts as soon as the two components are mixed. The paste–paste self-cured composite has a storage problem, as the BPO mixed with the dimethacrylate monomer is sensitive to heat decomposition. Therefore, inhibitors are added to suppress the polymerization of monomers and prevent premature polymerization. In the case of self-cured composites, inhibitors also provide clinical working time (referred to as the inhibition period, or more commonly, the induction period) to the professional for manipulating materials. When the two pastes of the composite are mixed, the radicals formed by the reaction of the BPO and amine are unable to react with the monomers due to the action of inhibitors and oxygen during the induction period. Thus, inhibitors produce an inert product, and completely stop polymerization until the inhibitors are consumed, after which polymerization proceeds at the same rate as if the inhibitor was absent [1]. Meanwhile, the material can be light-cured at any time during the chemical (self-) polymerization period. When the resin cement is irradiated, the production rate of primary radical is very high, so that the concentration of inhibitor decreases in a very short time, which explains the rapid decrease in amplitude of the NMR-MOUSE signal in Fig. 6a.
The mobile monomer concentration detected with the NMR-MOUSE decreases as the propagation stage of the reaction starts. The course of the NMR signal as a function of the curing time reveals that the beginning of the propagation stage in the self-cured sample is delayed compared to that of the photo-cured sample, due to the presence of the inhibitor in the cement composition. In this way, the induction period for the self-cured reaction is ~4 min (Fig. 6b). Although Fig. 6a shows an instant decay on the concentration of monomers, in this experiment, the induction period for the photo-cured reaction cannot be estimated.
The initial intensity of the CPMG decays decreases with growing distance from sensor for the photo-cured sample because of light absorption, such that the initial intensity for the profiles further down into the bulk of the sample is lower. During the dead time of measurement, a larger FR concentration was generated in the depth profile, leading to a higher polymerization rate, in accordance with Eq. (15), which states the time-and-depth dependence of the monomer concentration. As the depth into the sample increases, the polymerization rate decreases because of the poor transmittance through the resin cement mass. Studies have demonstrated that the transmittance through the restorative materials significantly influenced the degree of conversion and flexural strength [20]. In addition, opaque shades of the resin cement showed lower Knoop hardness than translucent shades for same irradiation time [21].
An expression for the changes in the mobile monomers concentration in the self-cure reaction was empirically obtained. During the inhibition time, it is assumed the rate of production of FRs to be identical to the rate of consumption of radicals by the inhibitors, because according to Fig. 6 there are no changes in the concentration of mobile monomer during the first minute. Thereafter, the monomer concentration decays in an exponential way. Considering the rate of self-polymerization in Eq. (9), we postulate this rate to be time-dependent:
in which \(K_{\text{SC}} = k_{\text{p}} \left( {k_{\text{d}} \frac{{(f_{1} + f_{2} )}}{{k_{\text{t}} }}} \right)^{{{\raise0.7ex\hbox{$1$} \!\mathord{\left/ {\vphantom {1 2}}\right.\kern-0pt} \!\lower0.7ex\hbox{$2$}}}} ([I][A])^{{{\raise0.7ex\hbox{$1$} \!\mathord{\left/ {\vphantom {1 2}}\right.\kern-0pt} \!\lower0.7ex\hbox{$2$}}}}\). This expression can be integrated to give the time-dependent monomer concentration,
where the initial time \(t_0\) is the average time for stopping the inhibition, \([M]_{0}\) the initial monomer concentration, and \(\tau_{\text{SC}} = (K_{\text{SC}} )^{{{\raise0.7ex\hbox{${ - 2}$} \!\mathord{\left/ {\vphantom {{ - 2} 3}}\right.\kern-0pt} \!\lower0.7ex\hbox{$3$}}}}\) the characteristic time constant of the self-cured reaction.
The model for the change in the monomers concentration for the self-cure (Eq. 17) has to be applied to the moment in time when the monomer concentration begins to decrease (Fig. 9a) after the inhibition time. The good agreement between theory and experiment supports the proposed model for the monomer concentration, although minor differences between model and experimental data can be observed at about 7.5 min. Some fluctuations can be observed at about 14.5 min in the experimental data, which can be attributed to the sensitivity of the sensor/noise. In addition, the model for the change in the monomers concentration for the photo-cure (Eq. 15) was applied. Figure 9b shows the fitted data for the 7.1 mm profile.

The entire curing process of a dual-cured resin without light irradiation is divided into three stages: the necessary working time for the dentistry professional; the conversion of monomers (curing time); and the cured sample (Fig. 7a). The EPR signal intensity for the self-cured sample first increases slowly during the time interval in which the monomers concentration is stable because of the presence of inhibitors to prevent premature reactions. In addition, oxygen trapped in the sample pans can act as an inhibitor [12]. Then, the intensity rapidly decays once the concentration of inhibitors is zero. At this point, the FRs can react with monomers and start the polymerization reaction. The EPR signal remains zero during the curing time of the resin cement because they are consumed at the same rate as they are generated, as assumed in the solution for the rate equations. High initial initiator concentrations are used in commercial polymerizations in order to achieve fully cured products in short reaction times [12, 22]. As the concentration of initiators is too large, FRs continue to be generated even after the monomers are converted into polymers (Fig. 7b), so the EPR signal intensity still grows after the curing time.
For the photo-cured sample, the amount of paramagnetic species detected in EPR spectroscopy increases during the irradiation time, and decreases after stopping irradiation (Fig. 8). The concentration of FRs rapidly increases up to a maximum value depending on the concentration of initiators, concentration of monomers, and other factors [23, 24]. When irradiation is terminated, the concentration of FRs decreases exponentially due to the consumption of FRs when converting monomers to polymers [4, 23–25]. Studies has demonstrated that FRs can be detected in a photopolymerizable dental resin long after stopping irradiation [24, 25]. The polymerization that occurs after irradiation is slow because the polymeric chain has low mobility in the matrix after irradiation.
The remaining FRs in a polymerized matrix have been widely studied [15, 24–28]. During radical polymerization, a gel effect occurs [26], increasing the concentration of FRs which cannot be terminated due to their reduced mobility in the matrix. In addition, a vitrification of the matrix occurs, and FRs [26] and remaining double bonds [27] are ‘quenched’ in the organic matrix [28]. Although FRs are present in the sample, the mobility of macromolecules is now in short range [24, 25, 28].
These results with real-time EPR spectroscopy can be related to the study with the NMR-MOUSE to confirm that the reduction in the concentration of mobile monomers traps FRs in the sample.
Conclusions
The measurements with the NMR-MOUSE showed that the mobile monomers concentration instantly decays for photo-cured samples, but it remains constant for ~4 min in the self-cured samples before starting to decrease. The experimental data were modeled with equations for first-order reaction kinetics in both polymerization protocols. The photo-cure of a dual-cured resin cement can be modeled as a function of reaction time and depth, and this process is independent of the self-cure, empirically modeled as function of reaction time.
Real-time polymerization monitoring by EPR for the self-cured sample showed that the concentration of free radicals suddenly decreases to zero after staying constant for ~4 min and then reappears with a highly increasing rate. These results apply to the three stages of the self-cured resin materials: (1) the necessary working time for the dentistry professional, (2) the chemical reaction by monomer addition to the chain (curing time), and (3) the vitrified (cured) sample, where there are no inhibitors or monomers available to react with the free radicals generated. For the self-cured sample, the concentration of free radicals increases strongly during irradiation, and starts to decrease immediately after irradiation.
These reported quantification of reaction kinetics and radical concentrations are relevant to dental materials research, because a detailed understanding the polymerization process can assist in improving resin cement formulations and curing procedures.
Abbreviations
- I :
-
Initiator
- A :
-
Amine
- M :
-
Monomer unit
- R :
-
Radical
- \({\text{RM}}_{n}^{ *}\) :
-
Active macroradical
- A * :
-
Amine radical
- I * :
-
Initiator radical
- \({\text{RM}}_{{n,{\text{b}}}}^{ *}\) :
-
Trapped macroradical with n monomer units
- Z :
-
Inhibitor
- D n+m :
-
“Dead” polymer containing n + m monomer units
- k d :
-
Initiator decomposition rate constant
- k p :
-
Propagation rate constant
- k t :
-
Termination rate constant
- k b :
-
Radical trapping rate constant
- k z :
-
Inhibition rate constant
- f 1 and f 2 :
-
Efficiency factors
- CQ:
-
Camphorquinone
- I 0 :
-
The intensity of light absorbed in the surface
- φ :
-
Extinction coefficient
- α :
-
Initiation efficiency
- d :
-
The length of the light path in the sample
- R PR :
-
Rate of production of primary radicals
- β :
-
Fraction of exciplex forming free radicals
- R P :
-
Rate of propagation
- R t :
-
Rate of termination
References
Kwon T-Y, Bagheri R, Kim YK, Kim K-H, Burrow MF (2012) Cure mechanisms in materials for use in esthetic dentistry. J Invest Clin Dent 3:3–16
Achilias DS, Sideridou I (2002) Study of the effect of two BPO/amine initiation system on the free radical polymerization of MMA used in dental resins and bone cements. J Macromol Sci A 39:1435–1450
Antonucci JM, Grams CL, Termini DJ (1979) New initiator systems for dental resins based on ascorbic acid. J Dent Res 58:1887–1899
Vicentin BLS, Salomão FM, Hoeppner MG, Di Mauro E (2016) Influence of geometrical configuration of a translucent fiberglass post on the polymerization of a dual cure resin cement analyzed by epr spectroscopy. Appl Magn Reson 47:211–222
Salomão FM, Vicentin BLS, Contreras EFR, Hoeppner MG, Di Mauro E (2015) The inflence of a translucent fiberglass post on the polymerization of dual cure resin cement analyzed by electron paramagnetic resonance. Mater Res 18:1023–1028
Silva ALF, Arias VG, Soares LES, Martin AM, Martins LRM (2007) Influence of fiber-post translucency on the degree of conversion of a dual-cured resin cement. J Endod 33:303–305
Calixto LR, Bandéca MC, Clavijo V, Andrade MF, Vaz LG, Campos EA (2012) Effect of resin cement system and root region on the push–out bond strength of a translucent fiber post. Oper Dent 37:80–86
Pegoraro TA, Da Silva NRFA, Carvalho RM (2007) Cements for use in esthetic dentistry. Dent Clin North Am 51:453–471
Fontes AS, Vicentin BLS, Valezi DF, Costa MF, Sano W, Di Mauro E (2014) A multifrequency (X-, Q-, and W-band) EPR and DFT study of a photopolymerizable dental resin. Appl Magn Reson 44:681–692
Netto AM, Steinhaus J, Hausnerova B, Moeginger B, Blümich B (2013) Time-resolved study of the photo-curing process of dental resins with the NMR-MOUSE. Appl Magn Reson 44:1027–1039
Blümich B, Haber-Pohlmeier S, Zia W (2014) Compact NMR. De Gruyter, Berlin
Achilias DS, Sideridou ID (2004) kinetics of the benzoyl peroxide/amine initiated free-radical polymerization of dental dimethacylate monomers: experimental studies and mathematical modeling for TEGDMA and Bis-EMA. Macromolecules 37:4254–4265
Vicentin BLS, Netto AM, Blümich B, Di Mauro E (2016) Identification of free radicals generated by different curing modes in a dental resin cement. Appl Magn Reson 47:1003–1014
Gear CW (1981) Numerical solution of ordinary differential equations: is there anything left to do? SIAM Rev 23:10–24
Sideridou ID, Achilias DS, Karava O (2006) Reactivity of benzoyl peroxide/amine system as an initiator for the free radical polymerization of dental and orthopaedic dimethacrylate monomers: effect of the amine and monomer chemical structure. Macromolecules 39:2072–2080
Truffier-Boutry D, Gallez XA, Demoustier-Champagne S, Devaux J, Mestdagh M, Champagne B, Leloup G (2003) Identification of free radicals trapped in solid methacrylated resins. J Polym Sci A Polym Chem 41:1691–1699
Watss DC (2005) Reaction kinetics and mechanics in photo-polymerised networks. Dent Mater 21:27–35
Hiemenz PC (1984) Polymer chemistry. Marcel Dekker, New York
Cook WD (1992) Photopolymerization kinetics of dimethacrylates using the camphorquinone amine initiator system. Polymer 68:125–151
Pick B, Gonzaga CC, Junior WS, Kawano Y, Braga RR, Cardoso PEC (2010) Influence of curing light attenuation caused by aesthetic indirect restorative materials on resin cement polymerization. Eur J Dent 4:314–323
Reges RV, Costa AR, Correr AB, Piva E, Puppin-Rontani RM, Sinhoreti MA, Correr-Sobrinho L (2009) Effect of light-curing units, post-cured time and shade of resin cement on Knoop hardness. Braz Dent J 20:410–413
Goodner MD, Bowman CN (1999) Modeling primary radical termination and its effects on autoacceleration in photopolymerization kinetics. Macromolecules 32:6552–6559
Fontes AS, Sano W, Dall’Antonia LH, Di Mauro E (2010) EPR in the characterization of the shade effect on translucence, remaining free radicals, and polymerization depth of commercially available resin composites. Appl Magn Reson 39:381–390
Leprince J, Lamblin G, Truffier-Boutry D, Demoustier-Champagne S, Devaux J, Mestdagh M, Leloup G (2009) Kinetic study of free radicals trapped in dental resins stored in different environments. Acta Biomater 5:2518–2524
Leprince JG, Lamblin G, Devaux J, Dewaele M, Mestdagh M, Palin WM, Gallez B, Leloup G (2010) Irradiation modes’ impact on radical entrapment in photoactive resins. J Dent Res 89:1494
Trommsdorff VE, Kole H, Lagally P (1948) Polymerization of methyl methacrylates. Makromol Chem 1:169–198
Ruyter IE, Svendsen SA (1978) Remaining methacrylate groups in composite restorative materials. Acta Odontol Scand 36:75–82
Truffier-Boutry D, Demoustier-Champagne S, Devaux J, Biebuyck JJ, Mestdagh M, Larbanois P, Leloup G (2006) A physico-chemical explanation of the post-polymerization shrinkage in dental resins. Dent Mater 22:405–412
Acknowledgements
The authors thank the FGM Produtos Odontológicos Ltda (Joinvile, SC, Brazil) for providing dental materials. The author AMN thanks Brazilian National Council for Scientific and Technological Development (CNPQ) for financial support (Grant No. 290010/2009-8).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vicentin, B.L.S., Netto, A.M., Dall’Antonia, L.H. et al. Real-time polymerization monitoring in a dual-cured resin cement by magnetic resonance. Polym. Bull. 74, 5163–5179 (2017). https://doi.org/10.1007/s00289-017-2007-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-017-2007-1