
Abstract
A series of blends of poly(vinyl alcohol)/poly(acrylic acid) (PVA/PAA) were prepared by solution mixing and casting. Glycerol was used as plasticizer. The blends were characterized for their physicochemical and thermo-mechanical properties. The FTIR results revealed the molecular level interaction between PVA and PAA at all blend ratios. The incorporation of PAA significantly reduced the storage modulus of PVA at a given temperature. PVA gradually lost its crystalline character with the increase of PAA and became fully amorphous when the PAA content in the blend exceeded 50 wt%. The kinetic parameters of the semi-crystalline blends were determined using the Avarami–Erofeev model, which showed excellent fitting with the experimental data from DSC. The loss in crystallinity of PVA also contributed to an increase in swelling of the blend when the PAA content is increased. The morphology study by FE-SEM demonstrated that there is no phase separation among the blend components at all blend ratios.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Extensive research is being carried out in academia as well as in industry to develop biodegradable polymer blends due to the concerns over conventional energy resources and increasing demands for advanced environment-friendly materials. Extreme attention is to be taken while replacing conventional polymers with biodegradable alternatives, because the fundamental mechanical, thermal and chemical properties have to be reproduced. The major features need to be analyzed for biodegradable polymers are water/solvent resistance, compatibility between blend components, blend ratio and special treatments required for specified properties.
Poly(vinyl alcohol) (PVA) is extensively used as water-soluble polymer in a wide range of applications such as drug delivery, water-soluble packaging films, oil field cementing, paper coating, textile sizing, hydrogels, etc. It possesses excellent properties like film formability, barrier to gases and liquids, hydrophilicity, adhesive strength, chemical resistance and more importantly its biodegradable nature [1–3]. The biodegradability is largely dependent on factors such as degree of polymerization, degree of saponification, tacticity of main chain, ethylene content and 1,2-glycol content [4]. PVA can be blended with different polymers or fillers to improve the properties and make use for different applications [5–8].
Poly(acrylic acid) (PAA or carbomer) is the basis of a class of materials called ‘super absorbent’. PAA can be used as polyelectrolyte with the ability to absorb and retain water and swell to many times its original volume. The swelling characteristics of PAA can be controlled by crosslinking reaction and copolymerization [9, 10]. PAA is used in many applications such as disposable diapers, ion exchange resins, toothpastes, controlled release devices, membranes and so on. PAA shows good miscibility with methyl cellulose polymer prepared by solution mixing and casting process. The subsequent thermal treatment leads to the formation of crosslinked material which is able to swell in aqueous and ethanol [11]. Recently, Alkan et al. [12] prepared poly(ethylene glycol)-PAA blend and found that at their miscibility conditions, the blends are potential material for thermal energy storage applications.
Poly(vinyl alcohol)/poly(acrylic acid) (PVA/PAA) is a well-known blend system for its molecular level miscibility through inter-polymer hydrogen bond interactions [13, 14]. They possess massive potential in bio/environmental-related applications as a biodegradable material because both of these polymers are water soluble and can be easily fabricated into insoluble swollen networks. The intermolecular ester groups formed between PVA and PAA are susceptible to microbiological degradation in the environment by soluble character of each component. From an engineering point of view, PVA can provide excellent strength properties and thermal stability, whereas PAA exhibits excellent swelling ability and ionic character. Quintero et al. studied the swelling behavior of PVA–PAA hydrogel films in solution of varying pH. They explained the possible application of this hydrogel as a bio-actuator/sensor based on its pH response [15]. PVA/PAA blends have been used in different applications such as the dehydration process of alcohols, ethanol or methanol [16] separation of organic solute molecules from water [17, 18], organic memory devices [19] and polymer electrolyte membranes for fuel cell applications [20]. Peppas and co-workers [21–24] have conducted extensive research on PVA/PAA blends to utilize them as hydrogel in drug delivery applications. The addition of PAA reduced the dry crystallinity of PVA, while the blend exhibited sensitivity to pH changes in the swelling solutions.
Our work, as such, aims to present comprehensive characterization of PVA/PAA blend at different blend ratios. The viability of using glycerol as a plasticizer in the blend is investigated in this study. The DSC cooling data are issued to produce the non-isothermal kinetic parameters using Avarami–Erofeev model.
Experimental
Materials
PVA (molecular weight, 27,000; degree of hydrolysis, 98.0–98.8 mol%) and glycerol were procured from Sigma-Aldrich Company. PAA (molecular weight, 1,250,000) was obtained from Winlab, UK. Ethanol (99.8 % absolute) of Sigma-Aldrich was used as solvent.
Preparation of PVA/PAA blends
The formulations are given in Table 1. Initially, specified amount of PVA as per the blend ratio was dissolved in deionized water at 90 °C and glycerol was added as the plasticizer. PAA was also weighed according to the blend ratio and was dissolved separately in ethanol at 70 °C. Ethanol was chosen as the solvent for PAA because PAA could be easily soluble in ethanol. When we used water as the solvent for PAA, much larger volume was required to dissolve same amount of PAA in water compared to ethanol as the solvent. This increases the processing complexity and evaporation duration. Once PVA and PAA were completely dissolved in water and ethanol, respectively, the PAA–ethanol solution was slowly added to PVA–water solution under constant stirring. The resulting mixture was then stirred for another 3 h to obtain a homogeneous solution and then poured into the glass Petri dish for drying at 50 °C for 24 h. The films were peeled off from the container and pressed between hot plates (at 150 °C) for 5 min using a Carver Press (Carver, Inc.) to obtain rectangular samples of 1 mm thickness (Fig. 1). The samples were preserved in a desiccator to avoid moisture uptake.

PVA/PAA films of different blend ratios after molding at 150 °C; a (90/10), b (70/30), c (50/50), d (30/70) and e (10/90)
Characterization
FTIR studies
The FTIR spectra were obtained by averaging 32 scans, at a resolution of 4 cm−1 from 500 to 4,500 cm−1 using a Thermo NICOLET 6700 FTIR Spectrometer.
Dynamic mechanical analysis (DMA)
Dynamic mechanical analysis (DMA) and properties of the samples were investigated in a temperature range from room temperature to 130 °C in tension mode at a heating rate of 10 °C min−1 and a frequency of 1 Hz using a Perkin Elmer DMA Q-800. The experiment was carried out under a nitrogen environment at a load of 5 N with an average sample size 4 × 10 × 1 mm.
Differential scanning calorimetry (DSC)
Differential scanning calorimetric (DSC) studies performed using DSC-Q1000, Universal V4.2E TA Instruments in nitrogen at a heating rate of 10 °C min−1 and in the temperature range −70 to 240 °C. DSC Calibration was completed by measuring the temperature and the enthalpy of indium melting. The percent crystallinity of PVA, PVA/starch blend and the composites was calculated from the ratio of the melting enthalpy of the sample to the theoretical melting enthalpy of 141.932 J g−1 of 100 % crystalline PVA, multiplied by the weight fraction of PVA in the sample [25].
Crystallization kinetics of PVA–PAA blends
The non-isothermal crystallization kinetics of PVA and PVA/PAA samples were evaluated using non-isothermal Avrami–Erofeev equation which is given by
where \({{f}}\left( {\alpha \left( {{T}} \right)} \right)\) is the non-isothermal Avrami–Erofeev crystallization function, α is the relative crystallinity, n represents the dimensions of crystal growth, k is the frequency factor, T 0 is the reference temperature and E a is the apparent activation energy. Detailed derivation of the equation can be found in Atiqullah et al. [26]. Equation (3) was solved by separation of variables into two functions α(T) and T (Eqs. 1 and 2). The non-linear model was then fitted to the experimental data using “NonLinearModelFit” command of Mathematica ®. The frequency factor was calculated at the reference temperature which was chosen as 370 K. Sufficient experimental data points were collected for estimating the kinetic parameters (k, E a and n) to ensure high accuracy.
α was calculated from the DSC exotherm by the formula given below: [26].
TGA analysis
Thermal degradation studies on samples weighing approximately 10 mg were performed using Perkin Elmer, Thermogravimetric Analyzer, Pyris-6 in nitrogen at a heating rate of 10 °C min−1 and in the temperature range 25–600 °C.
Measurement of equilibrium swelling
To investigate the swelling characteristics of the PVA/PAA blends in water, the rectangular-shaped samples were left to swell at room temperature. The swollen film was withdrawn from the solvent and quickly blotted with an absorbent paper to remove excess of superficial water, weighed and placed back in the same bath. The relative degree of swelling was measured at different time intervals until a constant weight was reached for each sample. This weight was used to calculate the equilibrium degree of swelling (S) using the following equation:
where W s and W d are the weights of the sample in the swollen and the dry state, respectively.
Gel content
To measure the gelation content, pre-weighed dry samples were immersed in deionized water and were allowed to equilibrium swelling. The samples were then taken out and dried in a vacuum oven at 50 °C until the weight of the sample was constant. The gel content was calculated using the following formula:
where W f is the final weight of the sample after swelling and drying and W i is the initial weight of the sample before the experiment.
FE-SEM studies
Cryo-fractured samples were gold coated and scanning electron micrograph (SEM) images were taken using LYRA3 TESCAN field emission SEM.
Results and discussion
FTIR studies
The FTIR spectra of PVA and PAA were compared to those of blends of varying compositions as given in Fig. 2. The molecular level miscibility between PVA and PAA could be achieved by the inter-polymer hydrogen-bonding interaction between the hydroxyl group of PVA and the carboxyl group of PAA. In pure PVA and PAA, the broad peaks at 3,000–3,500 cm−1 are ascribed to the hydroxyl groups. Pure PVA is a semi-crystalline material and the absorption region of –OH stretching (2,990–3,700 cm−1) is the contribution from the H bonds of crystalline and amorphous phases in PVA. More bandwidth of –OH group (2,700–3,700 cm−1) present in case of PAA is due to the intermolecular cyclic dimer interaction from the –COOH group in PAA. The hydroxyl group (–OH) stretching band of PVA at 3,280 cm−1 has weakened and broadened with the addition of PAA and tends to overlap with the broad band of carboxylic group (–COOH) of PAA in 2,400–3,500 cm−1 region. This indicates that the hydrogen bonds in PVA were steadily replaced by H bonds due to the interaction between PVA and PAA. The broad peak in PAA at 1,690 cm−1 is due to C=O (carbonyl) stretching vibration and stable intermolecular cyclic dimer. The peak resolution at 1,690 cm−1 reduced with increase in PVA content in the blend which indicates the formation of new intermolecular interaction between PVA and PAA at the expense of cyclic dimmers of PAA. The presence of water molecules bounded also contributes to the peaks in these broad regions mentioned above [27, 28]. The characteristic peak at 1,240 cm−1 corresponds to the –CH2– vibration of carboxylic group in PAA which becomes less intense due to the increase in polymer molecule chains’ rigidity as a result of interaction between PVA and PAA. The peak at 1,141 cm−1 is related to the C=O stretching in the crystalline region of PVA [29]. The crystalline structure of PVA disappeared completely when the PVA fraction is equal or less than that of PAA content in the blend. The gradual reduction in peak intensity and insignificant peak at higher concentration of PAA (above 50 wt%) indicates that the strong interactions established in the blend which hinder the formation of crystalline domains in PVA.

FTIR spectra of PVA, PAA and their blends at different compositions
Dynamic mechanical analysis (DMA)
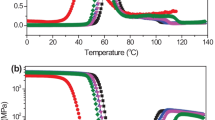
The elastic response (storage modulus) of PVA, PAA and their blends at varying compositions in a temperature range 40–130 °C is shown in Fig. 3. Compared to PAA, PVA is semi-crystalline and hence possesses higher mechanical strength properties. The incorporation of PAA into PVA caused significant decline in storage modulus. The blends show notable mechanical strength only up to a 70/30 composition of PVA/PAA. Equal weight ratio of PVA/PAA (50/50 composition) or higher PAA content blend (30/70 composition) produced a blend with considerably poor mechanical strength (Fig. 3). The molded films of PVA/PAA (90/10) were of irregular shape and too soft (Fig. 1e), so that the DMA study did not provide any useful information. Hence, it can be inferred that the storage modulus of the blends is basically governed by the contribution from the crystalline region of the PVA portion. As the PVA concentration in PVA/PAA blends drops, the strength contribution from the crystalline portion of PVA is reduced. The negligible crystallinity at 50 wt% or above of PAA concentration caused a drastic decrease in storage modulus.

Storage modulus vs. temperature of PVA and PVA/PAA blends
The storage modulus of PVA and the blends up to 70 wt% of PVA decreases with the increase in temperature within whole experimental range. However, in case of blends with higher amounts of PAA, after the initial drop in storage modulus, there was an increase in storage modulus at higher temperature above the T g of PAA (130 °C). This can be explained due to either or both of the following reasons. First, with the thermal effect, PAA can undergo crosslinking reaction, inter-molecularly between PVA and PAA by esterification reaction. Also, the intra-molecular cyclic anhydride formation in PAA due to elevated temperature can contribute some rigidity to the polymer chains (Scheme 1). The cyclic anhydrides in the PAA contributed more rigidity to the polymer chain, resulting in increase in storage modulus at higher temperature [30].

The chemical interaction between PVA and PAA via esterification reaction and the anhydride formation PAA
DSC studies
Figure 4 shows the heating and cooling thermograms of PVA, PAA and their blends at varying blend ratios. PVA shows clear melting and crystallization peaks whereas PAA does not show any, due to the semi-crystalline character of PVA and amorphous nature of PAA, respectively. In case of their blends, the melting and crystallization enthalpies decline with the decrease in melting and crystallization peak temperatures, as the PAA content in the total matrix is increased (Table 2). Both the melting and crystallization transitions disappeared completely for the blends with PAA concentration at 50 wt% or above. This clearly indicates that the crystallization process of PVA from the solution has been impaired by the presence of PAA. During the drying process of PVA/PAA blends, the PVA crystallites are formed simultaneously with the development of preferable interaction between PVA and PAA in the amorphous region of PVA. As the PAA content in the blend increased, the tendency of PVA crystallization fades because of the strong H-bonding interaction between PVA and PAA in the amorphous phase which impedes the PVA crystal formation.

DSC thermograms: the heating and the cooling curves of PVA, PAA and their blends
Crystallization kinetics of PVA–PAA blends
The non-isothermal crystallization kinetics of PVA crystallization at a cooling rate of 10 °C min−1 have been derived from the DSC data of crystallization regions of PVA, PVA/PAA (90/10) and PVA/PAA (70/30) blends (Eq. 3) [31]. Figure 5 shows the relative crystallinity profile for PVA, PVA/PAA (90/10) and PVA/PAA (70/30) blends predicted by the model in comparison with the experimental data. It is observed that the model predicts the crystallization profile very accurately with the coefficients of determination (R 2) above 0.995 for all samples. For the blends at or above 50 wt% of PAA, there was no crystallization curve and the model study was not used. The results from the model study are summarized in Table 3.

Model-predicted relative crystallinity vs. experimental data for PVA, PVA/PAA (90/10) and PVA/PAA (70/30) blends
The values of ‘n’ and ‘k’ for PVA are 3.1 and 0.0584 s−1, respectively, which shows that the crystal growth is tridimensional. These values are supported by the literature [32]. From the results shown in Table 1, it can be observed that the value of ‘n’ for the PVA/PAA blends is decreased which can be attributed to changes in crystal growth morphology [33]. However, the values for ‘k’ have increased even though there is significant reduction in the onset temperature of crystallization. The half-time of crystallization (i.e., time to complete 50 % crystallization after reaching crystallization onset temperature) was also reduced as the PAA content in the blend is increased. This indicates that crystallization process in PVA portion became faster even though there was marked decrease in total crystallinity of the blend [32]. The apparent activation energy ‘E a’ is also decreased slightly with the addition of PAA, therefore contributing to the ease of crystallization [26]. However, at higher loadings of PAA, the crystallization process is hindered by the excess amount of PAA.
TGA studies
The thermal stability of PVA, PAA and their blends at different ratios was studied using TGA and the results are shown in Fig. 6a, b. Compared to PVA, PAA is thermally more stable and its rate of degradation is lower. The maximum degradation peak temperature of PVA is at 298 °C whereas that of PAA is at 394 °C. The major degradation of PVA occurred in a temperature range 243–387 °C (ΔT = 144 °C). On the other hand, in case of PAA, the degradation took place over wide range of temperature between 195 and 490 °C (ΔT = 295 °C) with a much lower degradation rate. This is because the cyclic anhydride dimmers of the carboxylic group in PAA chains. They are thermally more stable than the intra-molecular H bonds in PVA.

a TGA and b DTG curves of PVA, PAA and their blends
In case of blends at all compositions, the initial degradation started at a lower temperature possibly due to the presence of glycerol which was added as plasticizer. The initial thermal degradation is also contributed by the cleavage of weak chemical bonds formed by the esterification reaction between PVA and PAA. However, as the PAA concentration in the blend increases, the major degradation peak was shifted to higher temperature, implying the improved thermal stability of the blend. The rate of degradation reaction of the blends in the major degradation zone is found to be in between that of PVA and PAA.
Equilibrium swelling studies
All the samples were swelled and reached equilibrium within 2 h, indicating that both PVA and PAA have fast swelling characteristics. It was also observed that, initially all the samples were swelled to a maximum and then there was a slight decrease in swollen weight with further increase in time. This is attributed to the dissolution of the loosely bound polymer chains into the solvent medium (water). In the case of blends, the swelling ratio increased with increase in PAA content in the blend due to the extensive swelling of PAA facilitated by the carboxylic acid groups on the polymer chain which strongly associated with water molecules [34]. If the blend components, PVA and PAA were considered individually, PAA possesses relatively higher swelling ratio compared to that of PVA (Fig. 7). This can be explained due to the presence of the crystalline domains in PVA which prevented the diffusion of water molecules into the polymer matrix. This indicates that the weight loss occurred due to leach-out from the amorphous portion of the material. There was a sharp increase in the degree of swelling at 50 wt% of PAA in the blend. At this concentration of PAA, the blend has lost its crystalline contribution from PVA, and completely behaved as an amorphous material. This is also supported by the DSC studies and previous literatures [13, 25]. There was more than 100 % swelling for the blends containing 50 wt% or above PAA.

Degree of swelling of PVA, PAA and their blends as a function of time
Gel content
The gel content of the samples measured after equilibrium swelling and drying demonstrated that the gel content increased with increase in PAA (58 % in case of PVA to 71 % for PAA) (Fig. 8). The tendency for PVA to leach-out and dissolve in water took place in its amorphous region. However, there was difference in the swelling response of PAA in water. In PAA swelling, water hydrates the weak acidic carboxylic groups attached to the polymer chain and the polymer go through stepwise swelling with ionization and neutralization of –COOH and –COO−, respectively. This can generate a steric interaction among the charged polymer groups and, in the long run, may lead to higher gel strength and swelling capability of the polymer. This is the reason why PAA absorbs more amount of water than PVA before the loosely bounded chains in the network dissolved in the given medium.

The degree of equilibrium swelling and gel content of PVA, PAA and their blends
FE-SEM studies
Cryo-fractured surfaces of the samples were examined under FE-SEM after etching the sample cross-sections using ethanol (Fig. 9). The PVA and PAA segments could make use of interaction by entanglement with the formation of intermolecular H-bonding and there is definite change in morphology as the blend ratio varies. The semi-crystalline character of PVA shows a ductile morphology with plastic deformation for the blends with rich PVA content [PVA/PAA (90/10)]. As the PAA content in the blend increased, the blend matrix has become more brittle and the fracture morphology became smoother. There was definite trend for the change in morphology as the PAA content increased from 10 to 90 wt%. The presence of tiny holes evenly right through the fractured surface for the blends with lower concentration of PAA indicates that the amorphous phase of PVA was leached out to ethanol solvent. The size of such hole reduced further with the increase of PAA content in the blend and finally disappeared as the PAA ratio reaches 90 wt% [i.e., PVA/PAA (10/90)]. At this composition, the blend shows nodular structure morphology. The absence of any phase separation or agglomeration for whole blend ratios indicates the excellent miscibility of PVA and PAA and the formation of a homogeneous blend at all the blend ratios.

FE-SEM photographs of PVA/PAA blends
Conclusions
Blends of poly(vinyl alcohol)/poly(acrylic acid) were prepared by solution mixing and casting. The use of glycerol as the plasticizer eased the blending process between PVA and PAA, and increased the compatibility of the final blend. The FTIR results showed the molecular level interaction between PVA and PAA at all blend ratios due to the hydrogen-bonding interaction. The incorporation of amorphous PAA polymer into semi-crystalline PVA significantly reduced the storage modulus of PVA at a given temperature. The loss in crystallinity for PVA was observed when the PAA concentration in the blend exceeds 50 wt%. The degree of swelling of the blends in water increased with increase in PAA in the blend whereas the dissolution of the blend components during equilibrium swelling period was decreased. PVA/PAA blends exhibit excellent miscibility at whole blend ratios as it is revealed from the absence of any phase separation or agglomeration. This study can be extremely useful considering the fact that biomaterial carrier for drug delivery applications needs to be crosslinked only physically so that there will not be any toxic leachable compounds or unreacted crosslinking agents.
References
IHS (2007) IHS chemical economics handbook, (polyvinyl alcohol). IHS, Englewood, USA
Baker MI, Walsh SP, Schwartz Z, Boyan BD (2012) A review of polyvinyl alcohol and its uses in cartilage and orthopedic applications. J Biomed Mater Res Part B Appl Biomater 100B:1451–1457. doi:10.1002/jbm.b.32694
Gupta B, Anjum S, Ikram S (2013) Characterization and physiochemical studies of crosslinked thiolated polyvinyl alcohol hydrogels. Polym Bull 70:2709–2725. doi:10.1007/s00289-013-0982-4
Kawai F, Hu X (2009) Biochemistry of microbial polyvinyl alcohol degradation. Appl Microbiol Biotechnol 84:227–237. doi:10.1007/s00253-009-2113-6
Tang X, Alavi S (2011) Recent advances in starch, polyvinyl alcohol based polymer blends, nanocomposites and their biodegradability. Carbohydr Polym 85:7–16. doi:10.1016/j.carbpol.2011.01.030
Santos C, Silva CJ, Buettel Z, Guimaraes R, Pereira SB, Tamagnini P, Zille A (2014) Preparation and characterization of polysaccharides/PVA blend nanofibrous membranes by electrospinning method. Carbohydr Polym 99:584–592. doi:10.1016/j.carbpol.2013.09.008
Singh R, Kulkarni SG, Shrikant S, Channe (2013) Thermal and mechanical properties of nano-titanium dioxide-doped polyvinyl alcohol. Polym Bull 70:1251–1264. doi:10.1007/s00289-012-0846-3
Zhu Y, Hu J, Wang J (2014) Removal of Co2+ from radioactive wastewater by polyvinyl alcohol (PVA)/chitosan magnetic composite. Prog Nucl Energy 71:172–178. doi:10.1016/j.pnucene.2013.12.005
Jabbari E, Nozari S (2000) Swelling behavior of acrylic acid hydrogels prepared by gamma-radiation crosslinking of polyacrylic acid in aqueous solution. Eur Polym J 36:2685–2692. doi:10.1016/S0014-3057(00)00044-6
Gao X, He C, Xiao C, Zhuang X, Chen X (2013) Biodegradable ph-responsive polyacrylic acid derivative hydrogels with tunable swelling behavior for oral delivery of insulin. Polymer 54:1786–1793. doi:10.1016/j.polymer.2013.01.050
Khutoryanskiy VV, Cascone MG, Lazzeri L, Nurkeeva ZS, Mun GA, Mangazbaeva RA (2003) Phase behaviour of methylcellulose–poly(acrylic acid) blends and preparation of related hydrophilic films. Polym Int 52:62–67. doi:10.1002/pi.1004
Alkan C, Guenther E, Hiebler S, Himpel M (2012) Complexing blends of polyacrylic acid–polyethylene glycol and poly(ethylene-co-acrylic acid)–polyethylene glycol as shape stabilized phase change materials. Energy Convers Manag 64:364–370. doi:10.1016/j.enconman.2012.06.003
Zhang XQ, Takegoshi K, Hikichi K (1992) Phase-separation and thermal-degradation of poly(vinyl alcohol) poly(methacrylic acid) and poly(vinyl alcohol) poly(acrylic acid) systems by c-13 cp/MAS NMR. Polymer 33:718–724. doi:10.1016/0032-3861(92)90327-S
Chen Q, Kurosu H, Ma L, Matsuo M (2002) Elongation-induced phase separation of poly(vinyl alcohol)/poly(acrylic-acid) blends as studied by c-13 cp/mas NMR and wide-angle X-ray diffraction. Polymer 43:1203–1206. doi:10.1016/S0032-3861(01)00690-5
Quintero SMM, Ponce RVF, Cremona M, Triques ALC, d’Almeida AR, Braga AMB (2010) Swelling and morphological properties of poly(vinyl alcohol) (PVA) and poly(acrylic acid) (PAA) hydrogels in solution with high salt concentration. Polymer 51:953–958. doi:10.1016/j.polymer.2009.12.016
Rhim JW, Lee SW, Kim YK (2002) Pervaporation separation of water-ethanol mixtures using metal-ion-exchanged poly(vinyl alcohol) (PVA)/sulfosuccinic acid (SSA) membranes. J Appl Polym Sci 85:1867–1873. doi:10.1002/app.10735
Rhim JW, Sohn MY, Lee KH (1994) Pervaporation separation of binary organic aqueous liquid-mixtures using cross-linked PVA membranes 2. Phenol water mixtures. J Appl Polym Sci 52:1217–1222. doi:10.1002/app.1994.070520906
Gudeman LF, Peppas NA (1995) pH-sensitive membranes from poly(vinyl alcohol) poly(acrylic acid) interpenetrating networks. J Membr Sci 107:239–248. doi:10.1016/0376-7388(95)00120-7
Ayesh AI, Qadri S, Baboo VJ, Haik MY, Haik Y (2013) Nano-floating gate organic memory devices utilizing Ag–Cu nanoparticles embedded in PVA–PAA–glycerol polymer. Synth Met 183:24–28. doi:10.1016/j.synthmet.2013.09.018
Kim DS, Park HB, Rhim JW, Lee YM (2005) Proton conductivity and methanol transport behavior of cross-linked PVA/PAA/silica hybrid membranes. Solid State Ionics 176:117–126. doi:10.1016/j.ssi.2004.07.011
Gudeman LF, Peppas NA (1995) Preparation and characterization of pH-sensitive, interpenetrating networks of poly(vinyl alcohol) and poly(acrylic acid). J Appl Polym Sci 55:919–928. doi:10.1002/app.1995.070550610
Hickey AS, Peppas NA (1997) Solute diffusion in poly(vinyl alcohol)/poly(acrylic acid) composite membranes prepared by freezing/thawing techniques. Polymer 38:5931–5936. doi:10.1016/S0032-3861(97)00163-8
Peppas NA, Wright SL (1998) Drug diffusion and binding in ionizable interpenetrating networks from poly(vinyl alcohol) and poly(acrylic acid). Eur J Pharm Biopharm 46:15–29. doi:10.1016/S0939-6411(97)00113-6
Peppas NA, Tennenhouse D (2004) Semicrystalline poly(vinyl alcohol) films and their blends with poly(acrylic acid) and poly(ethylene glycol) for drug delivery applications. J Drug Deliv Sci Tech 14:291–297
Nishio Y, Haratani T, Takahashi T, Manley RS (1989) Cellulose-poly(vinyl alcohol) blends—an estimation of thermodynamic polymer–polymer interaction by melting-point depression analysis. Macromol 22:2547–2549. doi:10.1021/ma00195a097
Atiqullah M, Hussain I, Al-Harbi A, Fazal A, Hossaen A (2012) Crystallization kinetics of ethylene homopolymers: a new perspective from residual catalyst and resin molecular weight. J Polym Res 19:1–13. doi:10.1007/s10965-011-9797-x
Daniliuc L, Dekesel C, David C (1992) Intermolecular interactions in blends of poly(vinyl alcohol) with poly(acrylic acid) 1. FTIR and DSC studies. Eur Polym J 28:1365–1371. doi:10.1016/0014-3057(92)90277-9
Manavi-Tehrani I, Rabiee M, Parviz M, Tahriri MR, Fahimi Z (2010) Preparation, characterization and controlled release investigation of biocompatible pH-sensitive PVA/PAA hydrogels. Macromol Symp 296:457–465. doi:10.1002/masy.201051062
Ping ZH, Nguyen QT, Neel J (1989) Investigations of polyvinyl-alcohol) poly(n-vinyl-2-pyrrolidone) blends: 1. Compat Makromolekulare Chemie—Macromol Chem Phys 190:437–448. doi:10.1002/macp.1989.021900222
Herrera-Kao W, Aguilar-Vega M (1999) Storage modulus changes with temperature in poly(vinyl alcohol) PVA/poly(acrylic acid) (PAA), blends. Polym Bull 42:449–456. doi:10.1007/s002890050488
Atiqullah M, Hossain MM, Kamal MS, Al-Harthi MA, Khan MJ, Hossaen A, Hussain I (2013) Crystallization kinetics of PE-β-isotactic PMMA diblock copolymer synthesized using SiMe2(Ind)2ZrMe2 and MAO co-catalyst. AIChE J 59:200–214. doi:10.1002/aic.13806
Lee KJ, Lee J, Hong J-Y, Jang J (2009) Influence of amorphous polymer nanoparticles on the crystallization behavior of poly(vinyl alcohol) nanocomposites. Macromol Res 17:476–482. doi:10.1007/BF03218895
Probst O, Moore EM, Resasco DE, Grady BP (2004) Nucleation of polyvinyl alcohol crystallization by single-walled carbon nanotubes. Polymer 45:4437–4443. doi:10.1016/j.polymer.2004.04.031
Elliott JE, Macdonald M, Nie J, Bowman CN (2004) Structure and swelling of poly(acrylic acid) hydrogels: effect of ph, ionic strength, and dilution on the crosslinked polymer structure. Polymer 45:1503–1510. doi:10.1016/j.polymer.2003.12.040
Acknowledgments
The authors would like to acknowledge the Deanship of Scientific Research, King Fahd University of Petroleum and Minerals for financial assistance through project number DSR IN121016.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jose, J., Shehzad, F. & Al-Harthi, M.A. Preparation method and physical, mechanical, thermal characterization of poly(vinyl alcohol)/poly(acrylic acid) blends. Polym. Bull. 71, 2787–2802 (2014). https://doi.org/10.1007/s00289-014-1221-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-014-1221-3