
Abstract
The present investigation deals with the mechanical and morphological properties of binary polyamide 12/maleic anhydride-grafted styrene-b-(ethylene-co-butylene)-b-styrene rubber (PA12/SEBS-g-MA) blends at varying dispersed phase (SEBS-g-MA) concentrations. Tensile behavior, impact strength and crystallinity of these blend systems were evaluated. Influence of microstructure, dispersed phase particle size, and ligament thickness on the impact toughness of the blend was studied. DSC data indicated an increase in crystallinity of PA12 in the blends. Tensile modulus and strength decreased while impact strength and elongation-at-break increased with the elastomer concentration. The enhanced properties were supported by interphase adhesion between the grafted maleic groups of rubber with polar moiety of polyamide 12. Analysis of the tensile data employing simple theoretical models showed the variation of stress concentration effect with blend composition.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nylons are engineering plastics possessing excellent property profiles, e.g., easy molding and extrudability, high strength and modulus, low coefficient of friction and wear, excellent chemical resistance, and high melting point [1–3]. Nylons may also be considered tough since they exhibit high-impact strength in falling dart tests and possess high work of fracture describing large area under the stress–strain curve [4]. Due to these properties, nylons find a wide variety of applications, e.g., in bearings, gears, motor housing, molded objects, fasteners and the likes. For still high-end applications nylons are rather brittle and notch sensitive at high strain rates and/or at low temperatures. Incorporation of an appropriate thermoplastic elastomer has been used extensively to counteract such embrittlement of nylons [5–10]. The elastomers usually contain acid, anhydride, or other functional groups. Maleic anhydride-grafted ethylene-propylene rubber (EPM-g-MA, EPDM-g-MA), styrene–ethylene–butylene–styrene thermoplastic elastomer (SEBS-g-MA) and also core shell rubbers made by emulsion polymerization methods are extensively used with nylons 6 [11, 12] and nylon 66 [13, 14]. The acid/anhydride/or other functional groups react with the amine end group and/or amide linkage producing nylon grafts on the elastomer [15]. It has been reported that styrene-assisted melt grafting enhances degree of grafting on impact modifier [16]. The decrease in the interfacial tension prevents coalescence of rubber particles through steric stabilization to give stable morphology and uniform dispersion of dispersed rubber particles in the polymer matrix [17, 18]. The graft structure promotes the phase adhesion and facilitates stress transfer between the phases. Several studies have documented that the size, distribution, concentration, interparticle ligament thickness and spatial arrangement of dispersed particle have profound effects on toughening efficiency of these impact modifiers [5, 10, 19]. From these studies it has been concluded that for toughening of the blend, size of the dispersed particle should be smaller than a critical value which depends on the concentration of the elastomer. Wu has proposed a single parameter, i.e., interparticle distance (ligament thickness T), which is independent of rubber concentration for toughening of a blend. The average value of ligament thickness should be lower than a critical value to impart toughness in the blend. Wu studied the rubber-modified polyamide 66 and established that the blends are toughened if the interparticle distance is <0.3 μm [5].
Shear yielding and cavitation in the rubber or matrix are thought to be primary energy-absorbing mechanism in rubber toughening of nylons [19–22] although crazing [23, 24], and fibrillation [25] within nylons have also been reported. Formation of hole is thought to be the critical requirement which precedes shear band formation. It has been observed that throughout the deformation process holes are formed in the rubber particles which are aligned along the shear bands. For easy shear deformation in the ductile mode and stable crack propagation through repeated loading to continuous energy absorption [8, 10], the rubber particle should be small so that cavitations are facilitated. Under this condition the triaxial tension in the deformation zone is dissipated facilitating plain-strain to plain-stress transition.
Polymer PA12 is a semi-crystalline nylon with low-equilibrium moisture absorption, excellent dimensional stability, very good chemical resistance and moderately high tensile and flexural strength. The polymer finds application in precision moldings for engineering, automobiles, cable and pump components [26]. Although PA12 has good to excellent impact resistance, incorporation of acid/anhydride containing elastomer increase the impact resistance with a minimum trade-off in other properties [27].
In this article, effect of incorporation of maleated styrene–ethylene–butylene–styrene (SEBS-g-MA) copolymer into PA12 has been studied at SEBS-g-MA concentrations 0 to 50 wt%. Tensile properties such as tensile modulus and strength and breaking elongation are evaluated and explained using models relevant to two-phase systems. Phase morphology was examined by SEM studies. The effect of particle size, particle size distribution and spatial arrangement is used to evaluate the impact strength of the blends.
Experimental
Materials
PA12 (Vestamid L 2140) used was a commercial product from Evonik Degussa High Performance Polymers, Germany. The melt-volume-flow rate (275 °C, 5 kg load) and density of PA12 were 36 cm3/10 min and 1.01 g/cm3, respectively. SEBS-g-MA with 1.4–2.0 wt% MA graft ratio and MFI of 22 g/10 min (230 °C, 5 kg load) was obtained from Kraton Polymers. SEBS-g-MA (specific gravity of 0.91 g/cm3) contained a styrene/rubber ratio 30/70 %, while number average molecular weights (M n) of the PS and poly (ethylene-co-butylene) copolymer were of approximately 7,000 and 37,500 g/mol, respectively [28].
Preparation of PA12/SEBS-g-MA blends and test specimens
PA12 and SEBS-g-MA pellets were dehumidified in a vacuum oven at 80 °C for 18 and 12 h, respectively. Melt mixing of the components was carried out on a co-rotating twin-screw extruder, Clextral, 900 mm (L/D = 42) at a screw speed of 300 rpm. Compounding was done when the torque stabilized at 75 ± 2 Nm. The extrusion zone temperature ranged from 220 to 240 °C. Thick strands leaving the extruder die were water cooled, dried and chopped into granules which were subsequently dried in vacuum at 80 °C for 12 h. The blends were injection-molded on a Krauss Maffei (CX series) injection-molding machine, into standard tensile bars with 4 mm thickness according to ISO 3167 test procedure. The melt and mold temperatures were maintained at 220–240 and 40 °C, respectively. The blends were designated as NS5, NS10, NS20, NS35, and NS50 containing 5, 10, 20, 35 and 50 wt% of SEBS-g-MA co-polymer.
Blend characterization
Internal morphology
The microstructural phase morphology of the blends was observed with cryo-fractured tensile specimens using a QUANTA 200 FEG environmental scanning electron microscope (FEI Company, System microanalyse Oxford Instrument, INCA INERGY 350, France). To prevent degradation by the electron beam, samples were coated with carbon using a Carbon Evaporation Device CED030 (Balzers). Energy dispersive SEM (ESEM) experiments were performed at a very low pressure. The samples were etched in toluene to remove SEBS-g-MA particles to evaluate elastomer particle diameter and extent of dispersion. The distribution of elastomer particles was obtained by measuring at least 150 particles. The number average diameter D n, weight average diameter D w, surface to volume average diameter D vs, and volume average diameter (D v) were determined by an automatic Image J analyzing technique using Eqs. (1–4):
where n i is the number of particles with apparent diameter D i (μm).
The interfacial area per unit volume of the blend (A i ) was calculated using Eq. (5):
where ϕ d is the volume fraction, and d is the average diameter of the dispersed phase, respectively.
The interparticle distance, called matrix ligament thickness (T) was calculated using theoretical equation given by Wu and Liu [5, 29–32]. Wu obtained a relation for ligament thickness by assuming that the dispersed particle occupies a definite lattice in the matrix, (Eq. 6) [5]:
where k = 1 for simple cubic (scc), k = 21/3 is for body-centered cubic (bcc) and k = 41/3 for face-centered (fcc) lattice arrangement of dispersed particles. In case of polymer blends with dispersed rubber particles, the distribution parameter, σ, accounts for the heterogeneity in the particle sizes is given by Eq. (7) [30]:
Wu modified the equation for ligament thickness by considering polydispersity of the distribution parameter (Eq. 8) [29]:
A modified equation by considering particle size, size distribution parameters of particles in the matrix was proposed by Liu et al. [31] (Eq. 9):
It has been established that the dispersed phase is randomly distributed in the polymer matrix as a result of processing. The true spatial distribution (ζ) of the dispersed particle depends on phase morphology of the blend. The average ligament thickness based on stacking configuration was calculated using Eq. (10):
Liu and his co-workers have noted that for most of the polymeric system spatial distribution parameter varied from 1.16 to 1.21 [12, 31, 32].
Fourier transform infra red spectroscopy (FTIR)
Fourier transform IR (FTIR) analysis of thin films of the blend components was carried out at a resolution of 2 cm−1 with wavelength ranging from 500–4,000 cm−1 on a Thermo Nicolet IR200 spectrometer. Thin film samples were prepared by compression molding machine at 220 °C and a pressure of 15 klb.
Mechanical property investigations
The test samples were first conditioned at an ambient temperature (22–25 °C) under 2–5 % relative humidity for 48 h before testing. Maximum tensile strength and elongation at break were measured using dumb-bell shaped specimens according to the ISO 527 standard test procedure at a crosshead speed of 50 mm/min using a Zwick Z100 tensile testing machine (load cell range of 0 to 10 kN). The values recorded were the averages of at least five specimens. Young’s modulus was also recorded using ISO 527 standard test procedure at a crosshead speed of 1 mm/min. Notched Charpy impact strength was determined using a Zwick 5102 apparatus according the ISO 179 standard test procedure (4 J). Testing was done on specimens of 80 × 10 × 4 mm3. As given by the manufacturer, root radius of the notch was 0.24 ± 0.05 mm, the notch angle was 45° ± 1° and the notch depth was 2.0 mm.
Crystallization studies
Crystallization behaviors of PA12 and PA12/SEBS-g-MA blends were studied by differential scanning calorimetry (DSC) on a Perkin Elmer Pyris Diamond DSC in an atmosphere of liquid nitrogen. The samples obtained by scraping small chips from the injection-molded samples were dried in a vacuum oven at 80 °C for 12 h before starting the experiment. Thermograms were recorded during the heating cycle at a constant heating rate of 10 °C per min. The heat of fusion values were used to calculate the percent crystallinity of PA12 in the blends using Eq. (11):
where \(\Delta H_{\text{m}}\) is the melting enthalpy of the sample, \(\Delta H_{{{\text{m}}_{0} }}\) the melting enthalpy of 100 % crystalline polymer (95 J/g) [28], and w the weight fraction of PA12.
Results and discussion
Microstructural phase morphology
Properties of blend mainly depend on the microstructural phase morphology of the system. Figure 1 shows the morphology of PA12/SEBS-g-MA blends where SEBS-g-MA copolymer is dispersed in polyamide matrix at all the compositions studied. Particle droplet (spheroidal) type morphology was observed with lower concentration of SEBS-g-MA (Fig. 1b–d), whereas mixed spheroidal, elongated and co-continuous phase was seen in NS35 (Fig. 1e). As the blend includes higher content of SEBS-g-MA in NS50, a well-developed co-continuous phase structure was observed (Fig. 1f). It can be noted that NS20 blend exhibits finer, uniform and stable phase morphology (Fig. 1d). The morphological parameters calculated by Image J analysis are presented in Table 1. The weight average particle diameter (D w) decreases from 0.48 μm (NS10) to 0.20 μm (NS20), further it increases with 35 and 50 wt% of elastomer which may be due to polymeric coalescence of the rubber phase at processing conditions as was also observed in other references [13, 14]. At 5 wt% of SEBS-g-MA (NS5), the elastomeric phase was not able to etch out with toluene which restricted the use of particle size analysis at this level (Fig. 1b).

Frequency distribution and SEM photomicrographs of cryogenically fractured etched surface of a N12; b NS5; c NS10; d NS20; e NS35; f NS50, respectively
In case of NS5, the amount of SEBS-g-MA was so small that most of the elastomer particles adhered to available polar groups of PA12 and was not removed by etching. As we moved toward 10 wt% of SEBS-g-MA (NS10) although the amount of grafted MA has increased as compared to NS5, but still the amount is not sufficient for very good polar–polar contact with PA12. Crystallizable PA12 also rejected some of the rubber particles in NS10 and hence bigger particles were seen. In NS20, the concentration of MA moiety of SEBS-g-MA appears to be comparable to polar groups of PA12 so that sufficient phase interaction takes place reducing the particle size of the dispersed phase [2, 11, 13]. At higher concentration of rubber, coalescence dominates and resulted with co-continuous morphology (NS35, NS50) but at lower concentrations particle breakup gives finer and uniform morphology as in the case of NS20 [27]. The lowering of particle size can be attributed to induced phase reactions between PA12 and maleic anhydride moiety which prevent the coalescence of the particles and finally lead to finer, stable and uniform morphology.
The distribution of dispersed particles can be evaluated from polydispersity index (D w/D n), Table 1, and frequency distribution curve shown in Fig. 1c–f. It is evident that in NS20, more finer size domains become prominent which leads to narrow width of the frequency distribution, Fig. 1c. This implies good mixing and high level of homogenization. However, NS50 blend shows the broadest particle distribution with larger particles due to dominance of coalescence and flocculation [12], Fig. 1f. The value for interfacial area A i, which is a measure of interfacial thickness, is much higher in case of NS20 and NS35, Table 1. This again gives an indication of better compatibility of blend which can help in stress transfer (discussed later). Table 1 depicted slight increase in D vs and D v when volume fraction of dispersed phase (ϕ d) increases from 0.21 to 0.37, but when ϕ d increases from 0.37 to 0.52 there is a considerable increase in D vs and D v values, indicating that the contribution toward interfacial area increases, when SEBS-g-MA concentration increases. The increase in Ai is much higher in case of NS20 and NS35, due to sufficient reduction in particle size, indicating enhanced possibility of stress transfers.
Matrix ligament thickness
Matrix ligament thickness calculated by Wu and Liu equations for all the compositions is shown in Table 1. The theoretical equations are based on the assumptions that the dispersed particle in the polyamide matrix can occupy a definite lattice arrangement (scc, bcc, fcc). The calculated values of ligament thickness (T) for different spatial configuration are denoted as T scc, T bcc, T fcc for k = 1, k = 21/3, k = 41/3 following Eq. (6) and presented in Table 1. Also the matrix ligament thickness calculated by Liu equations (Eqs. 9, 10) denoted as T Liu (ζ = 1) and T’Liu (ζ = 1.1 and ζ = 1.2) is shown in Table 1. It is not possible to give the exact value of spatial stacking configuration (ζ), however, for most of the system particle stacking configuration, ζ, varies between 1 and 1.2. The matrix ligament thickness predicted by Liu is higher than those predicted by Wu. It can be seen that the matrix ligament thickness decreases as the content of SEBS-g-MA increases. This is attributed to the increased coalescence of the dispersed phase.
FTIR analysis
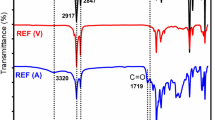
Figure 2 shows the FTIR spectra in the range of 4,000–500 cm−1 region for pure N12, SEBS-g-MA and their blends. SEBS-g-MA shows characteristic band at 1,868 and 1,745 cm−1 associated with symmetric and asymmetric vibrations of saturated five-membered anhydride ring (C=O stretching in maleic anhydride group) [33]. PA12 shows strong characteristic band at 3,290–3,091 cm−1 (N–H stretching), 2,954–2,921 cm−1 (C–H stretching), 1,638 cm−1 (C=O stretching, amide I band), and 1,560 cm−1 (N–H bending and C–N stretching, amide II band) [34, 35]. With the incorporation of SEBS-g-MA in PA12 matrix, amide I band shows a shift to lower frequency from 1,638 cm−1 (N12) to 1,632 cm−1 (NS5), 1,628 cm−1 (NS10), 1,631 cm−1 (NS20), 1,632 cm−1 (NS35), and 1,633 cm−1 (NS50), respectively. The PA12/SEBS-g-MA blends showed new bands at 1,740 cm−1 (NS5), 1,740 cm−1 (NS10), 1,741 cm−1 (NS20), 1,736 cm−1 (NS35), and 1,738 cm−1 (NS50), respectively, which have been assigned to imidization and grafting reactions between maleic anhydride of SEBS-g-MA and amine/amide group of polyamide 12 as depicted in Scheme 1 [2, 15, 36, 37]. Wu and his co-workers [38] reported that this interaction is responsible for transition of bare interfaces to occupied interfaces leading to lower interfacial tension.

FTIR spectra of N12, NS5, NS10, NS20, NS35, NS50 and SEBS-g-MA

Schematic representation of possible a amine-anhydride, b amide-anhydride reaction between Polyamide 12 and SEBS-g-MA
Degree of crystallinity
Crystallinity of a semi-crystalline thermoplastic material in a blend or composite is a function of the behaviors of the matrix as well as the dispersed phase. In some systems, the crystallinity decreases with increase in the content of the discrete phase because the latter interferes with the chain mobility of the former by creating a physical barrier [39, 40]. However, in other systems, the crystallinity enhances due to the inclusion’s nucleating activity through providing a surface for the crystallization of the polymer [41–43]. In PA12/SEBS-g-MA blends the crystallinity of the PA12 enhances with SEBS-g-MA concentration, Fig. 3. This may be due to the elastomer’s higher viscosity than PA12 at the processing temperature which facilitates nucleation of PA12. Since crystallinity of PA12 contributes to the mechanical properties of PA12/SEBS-g-MA blends, the parameter will be included in the description of the blend properties in the following sections.

Plot of crystallinity, X c, versus ϕ d in PA12/SEBS-g-MA blends
Tensile properties
Tensile strength and modulus and elongation-at-break of the blends were evaluated from the stress versus strain curve (not shown). In Figs. 4, 5, 6, these data are exhibited as plots of the ratios of the blend properties (subscript b) to that of PA12 (subscript m) versus ϕ d (the volume fraction of the dispersed phase SEBS-g-MA).



Plots of a relative elongation-at-break, є b/є m, of PA12/SEBS-g-MA blends and predicted behavior according to Mitsuishi model (Eq. 18). b Normalized relative elongation-at-break, [(є b/X c(b))/(є m/X c(m))], of PA12/SEBS-g-MA blends as a function of ϕ d
Tensile modulus
Plot of relative tensile modulus (E b/E m) versus ϕ d (Fig. 4) shows that the data decrease with increase in ϕ d. At the maximum ϕ d, the parameter decreased to ~0.5. This indicates that SEBS-g-MA copolymer softens PA12 substantially. The blend structure was evaluated using predictive models such as the “rule of mixture” [44, 45] for polymer blends/composites, Eq. (12), the “foam model” of Cohen and Ishai [46], Eq. (13), and “modified foam model” [47], Eq. (14):
where \(E_{\text{b}}\) and \(E_{\text{m}}\) denote the tensile modulus of the blend and the composites, respectively, \(E_{\text{d}}\) is the modulus of the dispersed phase SEBS-g-MA, and δ is the coefficient of foamability by the blending polymer. In the foam model, it is assumed that there is no phase interaction between the PA12 and SEBS-g-MA copolymer. The tensile moduli values for PA12 (E m = 1,464 MPa), and the blends (E b) were evaluated from the initial slopes of stress–strain curves using crosshead speed of 50 mm min−1, while the modulus value of SEBS-g-MA (E d = 187 MPa) was evaluated on compression module test samples. The rule of mixture curve shows higher values than the data, except at ϕ d = 0.05. It implies that although the dispersed phase provides a surface for crystallization, its flexibility possibly plays a dominant role in softening PA12. The relative moduli data were higher than the foam model, Eq. (13), Fig. 4a. The data showed good agreement with the modified foam model, Eq. (14), Fig. 4a. This indicates that SEBS-g-MA copolymer is not equivalent to a non-interacting pore/void, and there may be some kind of phase interaction through polar groups of PA12 and grafted maleic anhydride groups of SEBS-g-MA copolymer. However, since the MA concentration is very low, the extent of phase interaction is also limited.
The crystallinity of PA12 increased in the presence of SEBS-g-MA polymer which is also a flexible polymer. To evaluate the effect of these characteristics in the tensile modulus of the blends, the moduli were normalized by dividing the parameters by the crystallinity of PA12 in the blend and the matrix. In this way, the effect of crystallinity of the matrix was eliminated. The normalized relative moduli, \({{\left( {{{E_{\text{b}} } \mathord{\left/ {\vphantom {{E_{\text{b}} } {X_{\text{b}} }}} \right. \kern-0pt} {X_{\text{b}} }}} \right)} \mathord{\left/ {\vphantom {{\left( {{{E_{\text{b}} } \mathord{\left/ {\vphantom {{E_{\text{b}} } {X_{\text{b}} }}} \right. \kern-0pt} {X_{\text{b}} }}} \right)} {\left( {{{E_{\text{m}} } \mathord{\left/ {\vphantom {{E_{\text{m}} } {X_{\text{m}} }}} \right. \kern-0pt} {X_{\text{m}} }}} \right)}}} \right. \kern-0pt} {\left( {{{E_{\text{m}} } \mathord{\left/ {\vphantom {{E_{\text{m}} } {X_{\text{m}} }}} \right. \kern-0pt} {X_{\text{m}} }}} \right)}}\), plotted against ϕ d showed a continuous decrease, at the highest ϕ d, the value was ~0.3, Fig. 4b. Thus, the results showed that phase interaction, if any, is far suppressed by the flexibility of the blending phase SEBS-g-MA copolymer.
Tensile strength
The variations of relative tensile strength of PA12/SEBS-g-MA blends (ratio of tensile strength of PA12/SEBS-g-MA blend to that of PA12), (σ b/σ m) data versus ϕ d are presented in Fig. 5a. The tensile strength of PA12 decreased upon addition of SEBS-g-MA copolymer, the data varied between 0.86 and 0.54 depending on ϕ d, as a consequence of the elastomeric nature of the SEBS-g-MA which is responsible for weakening the PA12 matrix polymer structure. The weakening may be possibly due to a reduction of the effective matrix load-bearing cross-sectional area, similar to other elastomer-containing polymer blends [48]. Rubber particles act as stress concentrators promoting yielding in the interparticle ligaments and thus lower the yield stress of the blends. Using predictive models the relative tensile strength data were analyzed for weakness/discontinuity in the blend structure introduced by dispersed elastomer phase. Equation (16) describes Nicolais–Narkis Model [49, 50] whereas Porosity Model [51] is defined in Eq. (17):
These models assume the blend structure to be of a no-adhesion type and the tensile property as a function of either the area function or the volume fraction of the dispersed phase. It may be stated that the lower the value of K, Eq. (16), in the range 0–1.21 the better the phase cohesion. Similarly the weakness in structure or stress concentration is described by the parameter α, the higher the value the higher the extent of stress concentration.
The values of K and α for each ϕ d values are determined by comparing the experimental relative tensile strength data with the models [Eqs. (16) and (17)], and are shown in Table 2. The values of K are less than unity with an average value 0.88 which indicates that an extent of stress concentration exists in PA12/SEBS-g-MA blends. The extent of weakness or stress concentration is, however, low in the blend structure which may be due to a degree of phase interaction between maleic anhydride groups of SEBS-g-MA and the amine end group of PA12 [15]. The enhanced compatibility is achieved by possible interaction between the carbonyl oxygen in the maleic anhydride group of SEBS-g-MA and the hydrogen in the amide group of PA12.
Figure 5a exhibits comparison of the tensile stress values with Eqs. (16) and (17). The data agreed well with the Nicolais–Narkis model with an average K value 0.88 which implies a small extent of phase adhesion. With the Porosity model with an average of α = 1.93 the data also agreed well, the values show some scattering; the data were lower up to ϕ d = 0.1 and higher at ϕ d > 0.21.
Since PA12 is a semi-crystalline polymer, its crystallinity plays a role in its strength properties. The crystallinity of PA12 increased in the presence of SEBS-g-MA copolymer. There is also a kind of phase adhesion between the matrix polymer and the dispersed phase. SEBS-g-MA copolymer is, however, a flexible thermoplastic elastomer which softens PA12. To evaluate distinctively the role of crystallinity and phase adhesion vis-a-vis the elastomer flexibility, the normalized relative tensile strength values, \([{{(\sigma_{\text{b}} /X_{{{\text{c}}({\text{b}})}} )} \mathord{\left/ {\vphantom {{(\sigma_{\text{b}} /X_{{{\text{c}}({\text{b}})}} )} {(\sigma_{\text{m}} /X_{{{\text{c}}({\text{m}})}} )}}} \right. \kern-0pt} {(\sigma_{\text{m}} /X_{{{\text{c}}({\text{m}})}} )}}]\) were plotted as a function of ϕ d in Fig. 5b. The data were lower than unity varying from 0.83 to 0.4 at ϕ d values from 0.05 to 0.52. Thus, it may be considered that although SEBS-g-MA copolymer adheres to an extent with PA12 and provides a surface for crystallization of PA12, the elastomer flexibility predominates the former effects, bringing down the tensile strengths of the polyamide.
Elongation-at-break
Figure 6a exhibits that the relative elongation at break of the PA12/SEBS-g-MA blends increased continuously with ϕ d. The increase was from 42 to 234 % as ϕ d varied from 0.05 to 0.52, implying a substantial matrix softening by the rubbery dispersed phase. Tensile moduli and strengths values also indicated matrix softening by the discontinuous phase.
Modified Mitsuishi model [52] was employed to compare the elongation values, Eq. (18):
where ɛ b is the elongation at the break of the blends, ɛ m the value for the PA12, F the flexibility constant. The data exhibit quite good agreement with the model with an average F = 2.99, the values at ϕ d < 0.21 were lower while those at ϕ d > 0.21 were higher, all being almost evenly scattered around the curve.
The crystallinity of PA12 enhanced with ϕ d which should have increased the nylon’s rigidity. However, the ductility of the matrix enhanced despite the former effect which may be attributed to the predominant effect of softness of the elastomer. This increase in ductility of PA12 implies toughening of the polymer which will dissipate/absorb additional impact energy before failure, similar to other system [53].
Figure 6b presents a plot of relative normalized elongation at break data, \([(\varepsilon_{\text{b}} X_{{{\text{c}}({\text{b}})}} )/(\varepsilon_{\text{m}} /X_{{{\text{c}}({\text{m}})}} )]\), versus ϕ d. Here, the effect of enhancement of crystallinity of PA12 has been eliminated to evaluate the effect of blending SEBS-g-MA copolymer on the flexibility of PA12. The parameter increases up to ϕ d = 0.37 exhibiting a maximum of 2.66, the value decreased at ϕ d > 0.37 remaining higher than unity. However, the pattern of data variation validates the assumptions of the Mitsuishi et al. [52] model. This also indicates that SEBS-g-MA copolymer renders PA12 more deformable enhancing its impact toughness.
Impact properties
Figure 7a presents the plot of normalized relative notched charpy impact strength, (I b/X c(b))/(I m/X c(m)), of PA12/SEBS-g-MA blends against (ϕ d). With increasing concentration of impact modifier SEBS-g-MA, it was observed that impact strength of PA12 was increased by 39 to >500 % which may be attributed to the flexibility effect of SEBS-g-MA which enables the absorption of high-impact energy [27]. It is suggested that the modifier has toughened the PA12 by inducing energy dissipation mechanisms in the form of crazing and shear yielding process in the matrix. Such impact modification increases the softness and decreases the modulus of the blend, which give balanced property enhancement and could be used for further study with different filler for different end use applications. Further, the stiffness-normalized impact strength data (ratio of impact strength to Young’s modulus) which theoretically/semi-empirically express the toughness-to-stiffness combination has also increased with SEBS-g-MA content (Fig. 7b). Similar impact toughening was observed with other nylons [11, 54].

Plots of a normalized relative impact strength, [(I b/X c(b))/(I m/X c(m))], versus ϕ d. b Notched charpy impact strength (J/m) and impact strength/Young’s modulus ratio versus SEBS-g-MA concentration. c Variation in the impact strength with matrix ligament thickness
Structure–property relationship
The morphology evolved during melt processing of the blend has direct influence on the impact properties of the blend [29]. It has been reported that interparticle distance is one of the key parameters to evaluate the toughness of the blend. The variation in impact strength versus interparticle distance for PA12 and its blends is shown in Fig. 7c. It should be noted that NS50 blend did not break in the range of instrumental energy, indicated as non-breakable (NB). This indicates that the impact strength of the blends increases with decrease in interparticle distance. For NS20, the rubber particle size (0.20 µm) is neither too small nor too large which leads to effective toughening of the blend [7, 19].
The impact-fractured surface morphology of PA12/SEBS-g-MA blends is shown in Fig. 8. It is evident that stress whitening in the impact fractured surfaces is responsible for energy dissipation during impact testing of the blend. The impact energy applied to the blend is dissipated by rubber particles in the form of matrix yielding and crazing. Apart from this shear yielding may also be initiated at the stress concentration points which developed the local strain inhomogeneities [55]. Formation of such stress concentration points in the presence of SEBS-g-MA was indicated in the analysis of the tensile strength data. The shear bands also present a barrier to the propagation of crazes and hence crack growth, therefore, delaying the failure of the material.

SEM photomicrographs of impact fractured surfaces of a N12, b NS5, c NS10, d NS20 and e NS35
Conclusion
Flexibility of polyamide 12 has been increased with incorporation of SEBS-g-MA copolymer. Tensile modulus and strength decrease while ductility and toughness increase. SEBS-g-MA polymer provides a surface for the crystallization of PA12 enhancing its crystallinity but flexibility of SEBS-g-MA dominates the overall properties. The decrease in modulus and strength as well as increase in the elongation-at-break of the PA12 resulted from the softening effect of SEBS-g-MA which overrides the enhancement of crystallinity facilitating the molecular chain mobility of PA12.
The microstructure has controlled the properties of the blends which also depend on the processing conditions, while the work here only studied the performance of the final product at one condition (screw speed 300 rpm, temperature 220–240 °C). SEM observations indicated finer and globular morphology of the dispersed phase, as a result of particles breakup phenomenon at lower concentration of SEBS-g-MA and co-continuous morphology due to coalescence with higher elastomer contents. Significant increase in impact strength by about 1.3 to 7.3 times that of PA12 was observed. The blends showed ductile behavior, the ductility increases with decrease in interparticle distance. Tearing of rubber particles and the interphase caused stress whitening, which is responsible for the improved impact properties. The elastomer phase acts as a barrier to the crack propagation, therefore enhancing the failure energy of the blends.
References
Brydson JA (1999) Plastics materials, 7th edn. Butterworth Heinemann, Oxford, p 478
Keskulla H, Paul DR (1995) Toughened Nylons. In: Kohan MI (ed) Nylon plastics handbook. Hanser, New York, p 414
Welgos RJ (1998) Polyamide plastics. Encyclopedia of polymer science and engineering, vol 11, 2nd edn. Wiley, New York, pp 445–476
Utracki LA (2002) Polymer alloy and blends. Hanser, New York
Wu S (1985) Phase structure and adhesion in polymer blends: a criterion for rubber toughening. Polymer 26(12):1855–1863
Wu S, Margolina A (1990) Percolation model for brittle tough transition in nylon rubber blends-Reply to comments. Polymer 31(5):972–974
Borggreve RJM, Gaymans RJ (1989) Impact behaviour of nylon-rubber blends: 4. Effect of the coupling agent, maleic anhydride. Polymer 30(1):63–70
Gaymans RJ, Borggreve RJM (1989) Contemporary topics in polymer science: multiphase macromolecular systems, vol 6. Plenum, New York
Gaymans RJ, Dijkstra K (1990) Comments on ‘Percolation model for brittle-tough transition in nylon/rubber blends’. Polymer 31(5):971
Margolina A, Wu S (1988) Percolation model for brittle-tough transition in nylon/rubber blends. Polymer 29(12):2170–2173
Oshinski AJ, Keskkula H, Paul DR (1992) Rubber toughening of polyamides with functionalized block copolymers: 1. Nylon-6. Polymer 33(2):268–283
Balamurugan GP, Maiti SN (2007) Influence of microstructure and deformation behavior on toughening of reactively compatibilized polyamide 6 and poly (ethylene-co-butyl acrylate) blends. Eur Polymer J 43:1786–1805. doi:10.1016/j.eurpolymj.2007.02.035
Oshinski AJ, Keskkula H, Paul DR (1992) Rubber toughening of polyamides with functionalized block copolymers: 2. Nylon-6,6. Polymer 33(2):284–293
Muratoglu OK, Argon AS, Cohen RE, Weinberg M (1995) Toughening mechanism of rubber-modified polyamides. Polymer 36(5):921–930
Chow WS, Abu Bakar A, Mohd Ishak ZA, Karger-Kocsis J, Ishiaku US (2005) Effect of maleic anhydride-grafted ethylene–propylene rubber on the mechanical, rheological and morphological properties of organoclay reinforced polyamide 6/polypropylene nanocomposites. Eur Polymer J 41(4):687–696
Li Y, Xie X-M, Guo B-H (2001) Study on styrene-assisted melt free-radical grafting of maleic anhydride onto polypropylene. Polymer 42(8):3419–3425. doi:10.1016/S0032-3861(00)00767-9
Molau GE (1970) Colloidal and morphological behavior of block and graft copolymers. In: Aggarwal SL (ed) Block polymers. Plenum, New York, pp 79–106
Borggreve RJM (1988) Toughening of polyamide 6. PhD Thesis, University of Twente
Borggreve RJM, Gaymans RJ, Schuijer J, Housz JFI (1987) Brittle-tough transition in nylon-rubber blends: effect of rubber concentration and particle size. Polymer 28(9):1489–1496
Fukui T, Kikuchi Y, Inoue T (1991) Elastic-plastic analysis of the toughening mechanism in rubber-modified nylon: matrix yielding and cavitation. Polymer 32(13):2367–2371
Hobbs S, Dekkers M (1989) Deformation mechanisms in toughened poly(phenylene oxide)-polyamide blends. J Mater Sci 24(4):1316–1322. doi:10.1007/bf02397065
Bucknall CB (1990) Fracture resistance in rubber-toughened polymers. Makromol Chem Macromol Symp 38(1):1–15. doi:10.1002/masy19900380102
Narisawa I, Ishikawa M (1990) Crazing in semicrystalline thermoplastics. In: Kausch HH (ed) Crazing in polymers, vol 2. Springer, New York, p 375
Wu S (1983) Impact fracture mechanisms in polymer blends: rubber-toughened nylon. J Polym Sci Polym Phys Ed 21(5):699–716. doi:10.1002/pol1983-180210503
Michler GH (2008) High-impact rubber-modified polymers. In: Electron microscopy of polymers. Springer Laboratory. Springer, Berlin, pp 351–371. doi:10.1007/978-3-540-36352-119
Deng R (2002) Polyamide resins and their application, 1st edn. Chemical Industry Press, Beijing
Jose S, Thomas S, Lievana E, Karger-Kocsis J (2005) Morphology and mechanical properties of polyamide 12 blends with styrene/ethylene–butylene/styrene rubbers with and without maleation. J Appl Polym Sci 95(6):1376–1387. doi:10.1002/app21362
Jose S, Thomas PS, Thomas S, Karger-Kocsis J (2006) Thermal and crystallisation behaviours of blends of polyamide 12 with styrene–ethylene/butylene–styrene rubbers. Polymer 47(18):6328–6336
Wu S (1988) A generalized criterion for rubber toughening: the critical matrix ligament thickness. J Appl Polym Sci 35(2):549–561. doi:10.1002/app1988-070350220
Liu ZH, Zhang XD, Zhu XG, Qi ZN, Wang FS (1997) Effect of morphology on the brittle ductile transition of polymer blends: 1. A new equation for correlating morphological parameters. Polymer 38(21):5267–5273. doi:10.1016/s0032-3861-97-00075
Liu ZH, Li RKY, Tjong SC, Choy CL, Zhu XG, Qi ZN, Wang FS (1999) Influence of particle dispersion on the matrix ligament thickness of polymer blends: 2. A generalized equation and particle spatial distributions for different morphologies. Polymer 40(11):2903–2915. doi:10.1016/s0032-3861-98-00533-3
Liu ZH, Li RKY, Tjong SC, Qi ZN, Wang FS, Choy CL (1998) Influence of particle dispersion on the matrix ligament thickness of polymer blends. 1. The configuration of well-dispersed particles versus simple cubic lattice. Polymer 39(18):4433–4436. doi:10.1016/s0032-3861-97-10164-1
Kwee T, Mauritz KA, Beyer FL (2005) Poly[styrene-b-maleated (ethylene/butylene)-b-styrene] (mSEBS) block copolymers and mSEBS/inorganic nanocomposites: I. Morphology and FTIR characterization. Polymer 46(11):3871–3883. doi:10.1016/j.polymer.2005.02.076
Stuart B (2004) Infrared spectroscopy: fundamental and applications. John Wiley and Sons Ltd., Sussex
Garton A (1992) Infrared spectroscopy of polymer blends, composites and surfaces. Carl Hanser Verlag, Munich
Gaylord NG (1972) Compatibilization of hydroxyl-containing fillers and thermoplastic polymers. UK Patent
Kohan MI (1995) Nylon plastics handbook. Hanser, Munich
Wu C-J, Kuo J-F, Chen C-Y (1995) The role of the compatibilizer on morphologies and mechanical properties of polyamide 6/(styrene–[ethylene–butylene]–styrene) triblock copolymer blends. Adv Polym Technol 14(2):129–136. doi:10.1002/adv1995-060140204
Tomar N, Maiti SN (2007) Mechanical properties of PBT/ABAS blends. J Appl Polym Sci 104(3):1807–1817. doi:10.1002/app25831
Sewda K, Maiti SN (2007) Mechanical properties of HDPE/bark flour composites. J Appl Polym Sci 105(5):2598–2604. doi:10.1002/app26293
Kakkar D, Maiti SN (2012) Effect of flexibility of ethylene vinyl acetate and crystallization of polypropylene on the mechanical properties of i-PP/EVA blends. J Appl Polym Sci 123(3):1905–1912. doi:10.1002/app34680
Jafari SH, Gupta AK (1999) Crystallization behavior of polypropylene in polypropylene/nylon 6 blend. J Appl Polym Sci 71(7):1153–1161. doi:10.1002/(sici)1097-4628(19990214)71
Jafari SH (1999) Impact response of large specially orthotropic laminates. PhD Thesis, Indian Institute of Technology, New Delhi
Agarwal BD, Broutman LJ (1990) Analysis and performance of fiber composites. Wiley, New York
Maiti SN, Das R (2005) Mechanical properties of impact i-PP/CSM rubber blends. Int J Polym Mater 54(6):467–482
Cohen LJ, Ishai O (1967) The elastic properties of three-phase composites. J Compos Mater 1(4):390–403. doi:10.1177/002199836700100407
Choudhury A, Balmurulikrishnan A, Sarkhel G (2008) Polyamide 66/EPR-g-MA blends: mechanical modeling and kinetic analysis of thermal degradation. Polym Adv Technol 19(9):1226–1235. doi:10.1002/pat.1116
Palanivelu K, Sivaraman P, Dasaratha Reddy M (2002) Studies on thermoplastic polyurethane toughened poly(butylene terephthalate) blends. Polym Test 21(3):345–351
Nicolais L, Narkis M (1971) Stress–strain behavior of styrene-acrylonitrile/glass bead composites in the glassy region. Polym Eng Sci 11(3):194–199. doi:10.1002/pen.760110305
Kunori T, Geil PH (1980) Morphology-property relationships in polycarbonate-based blends. II. Tensile and impact strength. J Macromol Sci Part B Phys 18(1):1980
Nielsen LE (1967) Models for the permeability of filled polymer systems. J Macromol Sci Part A Chem 1(5):929–942. doi:10.1080/10601326708053745
Mitsuishi K, Kodama S, Kawasaki H (1985) Mechanical properties of polypropylene filled with calcium carbonate. Polym Eng Sci 25(17):1069–1073
Maiti SN, Barman N, Gupta AK (2005) Mechanical properties of ABS/CSM rubber blends. Int J Polym Mater 54(6):527–539. doi:10.1080/00914030390278635
Xie T, Yang G (2004) Effects of maleated styrene–(ethylene-co-butene)–styrene on compatibilization and properties of nylon-12,12/nylon-6 blends. J Appl Polym Sci 93(3):1446–1453. doi:10.1002/app.20588
Bowden PB (1970) A criterion for inhomogeneous plastic deformation. Phil Mag 22(177):455–462. doi:10.1080/14786437008225836
Acknowledgments
The authors gratefully acknowledge all the laboratory members for their generous help and support to one of the researcher Ms. Khushboo Rinawa. My special thanks to Mr. Belkacem Otazaghine, who provided guidance and support to Ms. Khushboo Rinawa during her stay in CMGD, laboratory in France. We are indebted to Mr. Marc Longerey for helpful assistance and discussions. We would also like to thank CSIR and Ecole des Mines d’Ales for providing funding and fellowship for carrying out this research work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rinawa, K., Maiti, S.N., Sonnier, R. et al. Influence of microstructure and flexibility of maleated styrene-b-(ethylene-co-butylene)-b-styrene rubber on the mechanical properties of polyamide 12. Polym. Bull. 71, 1131–1152 (2014). https://doi.org/10.1007/s00289-014-1115-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-014-1115-4