
Abstract
The copolymerization of styrene with cyclohexene, 1-methyl-1-cyclohexene, and norbornene using ethenylbisindenylzirconium dichloride and methylaluminoxane, Et(Ind)2ZrCl2-MAO, initiating systems has been tested. The results obtained with each styrene-cycloalkene couple, except styrene/norbornene, indicate a less effective polymerization process compared to styrene homopolymerization, in agreement with the electronic and steric effects present in each comonomer. The electronic I+ effects of substituent groups, depending on their placement, largely improve the polymerization process, while bulky groups on or near the vinyl carbon double bond of styrene decrease its effectiveness. The present study shows that the copolymers obtained are amorphous and their composition showed a lower abundance of comonomer units with respect to the initial feed. For comparison, the results of the copolymerization of styrene/(1-octadecene) using the same initiator system and polymerization process are included, a polymerization that indicates a more reactive process, and as the proportion of octadecene in the initial feed increases, it showed a crystalline fusion temperature as well as a Tg in the styrene region which can be attributed to the formation of block styrene/octadecene copolymers.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The homo- and copolymerization of styrene has been studied very extensively. Comonomers such as substituted styrenes, alkenes, and dienes have been studied most widely. The main objective of these studies has been to improve the properties of conventional polystyrene, one of the most largely used polymers because of its properties, low cost and easy manufacture. Some important reviews on the subject have also been published [1, 2]. In addition to these, the objectives have included the elucidation of the mechanism of metallocene-induced styrene polymerization.
Initially our efforts regarding styrene (S) polymerization were addressed at improving the temperature and resistance to organic solvents of conventional polystyrene, PS, particularly through the synthesis of stereoregular polystyrene, e.g., syndiotactic polystyrene, s-PS. Then we aimed at improving the flexibility of the final products, thereby increasing their impact resistance. It is known that phenyl groups along PS chains contribute to polymer rigidity, making them stiff and fragile. The idea was to incorporate comonomer units along the styrene polymer chains which would act as spacing agents of phenyl groups in the copolymer structure.
It is known that in the presence of some specific catalytic systems the cycloolefins (cycloalkenes = CyAlk) undergo vinyl and ring-opening metathesis polymerization, yielding poly(cycloalkene)s and polyalkenamers, respectively [3].
Generally, the vinyl polymerization reactions of cycloolefins are initiated by cationic and Ziegler–Natta coordination catalysts, while the ring-opening polymerization is promoted by metathesis (ring-opening monomer polymerization, ROMP) catalysts. Vinyl polymerization has found various applications in the manufacture of hydrocarbon resins [4] and recently in the production of copolymers of cycloolefins [7, 8]. Metathesis polymerization has become a versatile method for the synthesis of a large class of polymers having desired physical and chemical properties, particularly good mechanical, electrical, and optical characteristics, as well as superior weathering and heat resistance [5–8].
Cycloalkenes, unlike acyclic 1,2-disubstituted ethylenes, undergo Ziegler–Natta polymerization due to the presence of ring strain. Two polymerization routes are possible: polymerization through the double bond or by a ring-opening (olefin metathesis) reaction, for example, with cyclobutene. Each of the two polymerization routes can yield poly(1,2-cyclobutene) by vinyl polymerization which can exist as either the erythrodiisotactic or erythrodisyndiotactic isomer. The second one, metathesis, yields –(CH2CH=CHCH2)– which can exist as either the cis or trans isomer. Exclusive polymerization through the double bond does not occur with any other cycloalkene. It is reported that cyclohexene does not polymerize by either route in the presence of various initiators except when the cyclohexene structure is part of a bicyclic system like norbornene [9].
Hoffman reported the preparation of oligomers such as dimers, trimers, and tetramers, while Boor et al. [10] pointed out that cyclohexene with AlCl3 at low temperature (−20 °C) did not produce any polymer, suggesting that temperature plays a significant role in the polymerization process. Furthermore, the structure of the resulting polymer should be more complex as cyclohexyl cations can easily undergo 1,2-, 1,3- and 1,4-intramolecular changes under the action of strong Lewis acids, to generate carbocations more easily accessible to the enchainment of monomers during propagation reactions.
For substituted cyclohexene such as 1-methyl-1-cyclohexene, Roberts and Day [11] examined its polymerization with AlCl3 in benzene at 40–45 °C and obtained oligomer mixtures of 1-methyl-1-cyclohexene, mainly dimers. High polymers with 1,2-structures can be obtained under more severe conditions.
For cationic polymerization of norbornene, the authors indicate the production of poly(norbornene) or poly(1,2-norbornilene) through a 1,2-poly-addition to double bonded carbons. But under the action of Lewis acids on the carbonyl cation resulting in the initiation and propagation stages, hybrid shifts and rearrangements of the bicyclic skeleton are frequently produced.
In their research on Ziegler–Natta norbornene polymerization, Tsujino et al. [12] used various cationic catalysts: BF3·Et2O, AlCl3, and AlCl4, and got low molecular weight polymers; they also assumed that they were due to 2,3-rearrangements.
Kennedy and Markowski [13] reported on norbornene polymerization in the presence of EtAlCl2 in EtCl at low temperature, obtaining white, soluble, low molecular weight (1,470 and 1,940) polymers, but with high softening points, 235 and 260 °C, respectively. The authors assumed that the polymers contained 2,3-arrangements in the repeating unit. Later, Kennedy suggested that polynorbornene prepared by conventional cationic polymerization is a mixture of recurrent units resulting from isomerism of the norbornene skeleton “prior” to the polymerization process.
Deffieux et al.[14] report on the homo- and copolymerization of styrene with Ni-based/MAO catalysts, concluding that the fully saturated structure of polynorbornene indicates that the two monomers polymerize by an ethylenic type addition reaction and that the synthesis of true copolymers shows that one type of active species is operating for the two monomers, where determination of reactivity ratios shows a much higher reactivity of norbornene.
In studying styrene copolymerization we have tried various comonomers, first alkylsubstituted styrenes such as para-methylstyrene [15] and para-tert-butyl-styrene which yielded products with Tg higher than conventional PS [16], then 1-alkenes, particularly long-chain 1-alkenes such as 1-hexadecene and 1-octadecene [17, 18], and dienes such as 2-methyl-1,3-butadiene [19].
When working with a cyclopentadienyltitanium trichloride-MAO initiator system, homopolymerization of p-methylstyrene and, better yet, p-tertbutylstyrene, there was significant improvement in polymer yields compared with the homopolymerization of styrene. The copolymerization of such comonomers with styrene produced higher conversion according to the proportion of p-methyl or p-tert-butyl comonomer in the initial copolymerization feed. From such results, it was concluded that polymerization was much improved depending on the electronic effect of the substituent groups, whose inductive effect activated styrene’s vinyl group increasing its electron density and therefore facilitating monomer coordination with active initiator species. It was concluded that styrene polymerization was improved by the I+ electronic effect of appropriately positioned alkyl groups, while halogen substituted styrene—with the halogen having an I− inductive effect—decreases conversion to polymer. A similar situation occurred when substituent groups, regardless of their I+ or I− effect, were placed in a particular position, the ortho position on styrene and/or alpha methyl substitution on the vinyl group of styrene, decrease of polymerization efficiency, and a marked or total efficiency decrease was further established for 2,6-dimethyl- and 2,4,6-trimethyl-styrene [20]. This situation was attributed to steric hindrance, which makes the coordination of such monomer with the active species, and the subsequent propagation process, more difficult. We concluded that styrene polymerization through these metallocene-MAO initiator system is greatly influenced not only by electronic effects but also by steric hindrance [21]. So ortho-MeS and α-MeS turned out to be less prone than styrene to polymerize through the metallocene-MAO initiating systems.
The present paper reports on our recent studies of styrene-cycloalkene copolymerization using the initiator system ethenylbisindenylzirconium dichloride and methylaluminoxane, Et(Ind)2ZrCl2–MAO, which is more effective for 1-alkene polymerization than its titanocene counterpart. For comparison, in this paper we include copolymerization experiments of S/1-octadecene performed with the same initiator system and polymerization conditions used for the S/cycloolefin copolymerizations studies.
Experimental
The polymerization reactions, purification and handling of styrene monomer and solvents were done as reported previously [16, 20, 21], using Schlenk reactors and glove box techniques. Solvent toluene, MAO and metallocene solutions in toluene were charged sequentially by syringe under argon pressure. Polymerization was initiated by injecting either the styrene followed by the comonomer, or both of them simultaneously.
Differential scanning calorimetry (DSC) analyses were performed using a Rheometric Scientific or a Perkin Elmer Pris 1 DSC apparatus with 3 to 4 mg samples placed in a nitrogen atmosphere, heating them at a rate of 10 °C min−1, and after cooling to room temperature, reheating at the same rate. The reported Tg and Tm values were those recorded on the second heating scan.
1H-NMR spectra were recorded on a Bruker AVANCE-400 spectrometer at 60 °C, operating at 400.13 MHz. The polymers and copolymers were dissolved in deuterated chloroform or deuterated 1,1,2,2-tetrachloroethane. A total of 64 K data points with a relaxation delay of 1.5 s were collected. Chemical shifts were calibrated with respect to the residual protonated signal of the solvent (CDCl3, 7.26 ppm and C2D2Cl4, 6.0 ppm) and reported relative to tetramethylsilane (TMS). The copolymer composition was established from the integration values in the aromatic and aliphatic regions (see Table 1).
Results and discussion
Continuing with our studies on styrene copolymerization, we have attempted the copolymerization of S/cyclohexene, (CyHex), S/(1-methyl-1-cyclohexene, MeCyHex), and S/bicyclo[2.2.1]hept-2-ene, known as 2-norbornene, (Norb), or norbornylene.
Table 1 shows the results of the copolymerization of styrene with cycloalkene comonomers, where it is seen that the presence of both cyclohexene and 1-Me-cyclohexene decreases conversion to copolymer with respect to the homopolymerization of styrene. In other words, these comonomers decrease the efficiency of the polymerization process catalyzed by Et(Ind)2ZrCl2-MAO and they are less reactive than styrene itself. This situation may be related to the lower reactivity of the cycloolefins to generate poly(cycloolefin) polymers through vinyl polymerization. On the other hand, results of S/norbornene show increased copolymer yield compared to cyclohexene and 1-methyl-1-cyclohexene, pointing to a more reactive comonomer under the experimental conditions.
Furthermore, the results in Table 1 show low intrinsic viscosity values, in agreement with the low molecular weights of the resultant copolymers. Moreover, the composition of the products shows a lower proportion of cycloolefin than in the initial feed, in agreement with the low tendency of cyclohexene and substituted cyclohexene to undergo vinyl polymerization. Still, there is another factor that must be taken in account, which is the steric hindrance resulting from the incoming monomer. It is also important to consider the structure of the active species of the catalyst complex.
The rac-bisindenylzirconiumdichloride metallocene has two labile chloride atoms which are replaced by methyl groups in the presence of MAO, followed by an α-agostic interaction, where the zirconium is lacking electrons and is the place where the vinyl monomer will coordinate with the Zr+ (see Fig. 1), so the more bulky the incoming monomer is, either styrene or cycloalkene, the more difficult its complexing will be, so styrene, having a pendant CH2=CH– group, will lead more easily to the formation of such complex than the cycloolefin, where vinyl is part of the ring structure, and consequently a lower number of units of the latter will appear in the copolymer. Furthermore, considering that CyHex, MeCyHex, and Norb present different coordination capacities depending on the particular reactivity of the vinyl group, norbornene will be more abundant in the copolymer chain, followed by MeCyHex, which has a methyl group bonded to the vinyl portion. As the propagation process takes place through insertion of the incoming monomer, the incorporation of the following unit will be more difficult, after an immediately previous bulky cyclic alkene unit inserted in such growing polymer chain.

a α-Agostic association. b Approaching monomer unit to the “agostic” active species
Figure 1 illustrates the situation that arises when considering an α-agostic interaction in a likeness of the one presented by Macrogallery for cyclopentadienyl-zirconium-dichloride [23].
The α-agostic association tells about the interaction between hydrogen atoms of the methyl group of the active Zr+ species, to which the incoming monomer coordinate. Figure 1a shows the α-agostic association by which the positively charged zirconium is stabilized by electrons from the hydrogen of the ligand’s methyl. However, the zirconium is still lacking electrons. It needs just a weakly agostic association to satisfy it. That is where the olefin monomer comes in, as shown in b, when, as an example, a propylene incoming unit approaches. Such situation is further discussed by K.D. Karlin in his Organometallic Chemistry course [24] and also by M. Brookhart et al. [25, 26] in their publications on the impact of agostic interaction on the structure and reactivity of organotransition metal compounds.
Table 1 also shows the activity ratio, which has been calculated from the corresponding activity (kg copolymer molZr−1 mol(S+ comonomer)−1 h−1) of the S polymerization against the activity of the particular copolymer. That shows the effectiveness of the initiator system with respect to the particular S/comonomer couple when compared to the homopolymerization of styrene.
In a previous paper [27] related to the copolymerization of propylene/norbornene through the Si-bridged metallocene Me2Si(2-Me-Ind)2ZrCl2, higher conversions than that reported here were reached, so we consider that in the latter system the incoming monomer will have more room to reach and coordinate with the Zr+ as a consequence of the larger opening at the complexing place [28] because of the smaller angle generated by the Si bridge compared to the –CH2CH2– bridge of our metallocene. However, in that paper we also reported that the presence of norbornene decreases polymerization activity by ca. 80 % when it is fed in low proportion.
Figure 2 shows the 1H-NMR spectrum of styrene/norbornene copolymers prepared using a nickel(II) complex bearing acyl hydrazone ligands (bis[N-(pyridine-2-carboxaldehide)benzoylhydrazone]nickel(II) dibromide) in the presence of MAO. The shapes of these spectra are similar to those of our copolymers, which are shown in Fig. 3.

1H-NMR spectra of norbornene/styrene copolymers. (Taken from Ogata et al. [29]). “This figure is reproduced with permission from John Wiley & Sons, Inc.”

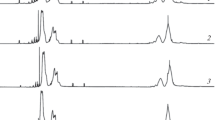
1H-NMR spectra in CDCl3 or C2D2Cl4 at 60 oC, of copolymers: a S/CyHex; b S/MeCyHex; c S/Norb; obtained with the Et(Ind)2ZrCl2-MAO initiator system in toluene after 48 h at 60 °C. S/CyAlk (mol/mol) ratios in the initial feed are indicated for each spectrum
The 1H-NMR spectra of S/cyclohexene copolymers (Fig. 3a) show the corresponding signals in both the aromatic and aliphatic regions. The corresponding copolymer compositions reported in Table 1 were calculated from their integrals. It is seen that for each copolymer composition, there is a lower incorporation of cycloalkene with respect to its proportion in the initial feed.
For S/1-methylcyclohexene, the NMR signals appear again in both regions (Fig. 3b), pointing to the incorporation of comonomer in the copolymer composition. Furthermore, in the aliphatic region the signal of the –CH3 substituent group of methylcyclohexene can be seen. As in the previous case, however, a lower proportion of comonomer with respect to the initial feed is incorporated in the copolymer.
Figure 3c shows the 1H-NMR spectra of the S/norbornene copolymers. In this case, there is a greater incorporation of comonomer with respect to its proportion in the initial feed, pointing to a behavior that differs from that of the other two cylohexene comonomers. So conversion to polymer increases as the proportion of norbornene increases in the initial feed, indicating that norbornene is more prone to polymerize, perhaps because its C–C double bond is more exposed to coordination with the active species. However, as already mentioned, norbornene is more reactive than cyclohexene and its derivatives [14].
As can be seen, particularly in the aromatic region of the NMR spectra, all the copolymers were amorphous. The DSC curves are also indicative of the amorphous nature of the copolymers.
Table 2 shows the results for styrene/1-octadecene copolymerization with the Et(Ind)2ZrCl2-MAO initiating system, showing a sustained improvement in copolymer conversion as well as increased 1-octadecene incorporation in the resulting copolymer, higher than the proportion of comonomer in the initial feed. This is an already established result for zirconocene-MAO initiator systems [17, 18], which are better than titanocene-MAO initiator systems for 1-alkenes.
The 1H-NMR spectra in Fig. 4 indicate the presence of both comonomers in the copolymer chains, confirming their nature as true copolymers. In Table 2, it is also clear that the resulting copolymers are richer in octadecene than its proportion in the initial feed, showing that 1-C8H16 is more prone to polymerize than styrene, as shown by the activity ratio for that copolymerization.

1H-NMR spectra of S/1-octadecene copolymers in CDCl3, obtained using an Et2(Ind)2ZrCl2–MAO initiator system in toluene, after 48 h at 60 °C
The DSC results, shown in Fig. 5, as well as the NMR spectra (Fig. 3), are also in agreement with the amorphous nature of the S/cycloalkene copolymers. Furthermore, the lower value of the glass transition temperature suggests that these copolymers should be less rigid than PS.

DSC thermograms of copolymers using the Et(Ind)2ZrCl2-MAO initiator system: a S/cyclohexene, b S/1-methyl-1-cyclohexene, c S/norbornene, d S/(1-octadecene), in toluene, after 48 h at 60 °C. S/comonomer (mol/mol) ratios in initial feed are shown for each spectrum
On the other hand, the DSC thermograms of the S/(1-octadecene) copolymer with initial 75/25, 50/50, and 25/75 mol/mol S/C18H36 feeds (Fig. 5d) show their crystalline nature, as had been established earlier for their copolymerization using Ph2Zn-Et(Ind)2ZrCl2-MAO initiator systems [17, 18].
The DSC thermograms of styrene/1-octadecene copolymers, particularly those with higher proportions of octadecene in the initial feed, show a melting fusion temperature which we attribute to ordered polymer chains due to pendant methylene segments –(CH2)15CH3 arising from octadecene units incorporated in an isotactic sequence, as was concluded for S/C18H36 polymerization catalyzed by Ph2Zn-Et(Ind)2ZrCl2-MAO [17]. In other words, the Tm and Tg signals mark both blocks and consequently the presence of a block P(S-co-octadecene) copolymer. Figure 6 is in agreement with this statement, since, as shown, it was possible to evaluate by DSC a Tg (77.0 °C) attributed to polystyrene with some occasional insertion of octadecene (defects in parts of the PS chain) and a Tm (26.6 °C) generated by the ordered pendant –(CH2)15CH3 groups of the incorporated 1-octadecene units (blocks).

DSC thermogram of the S/1-octadecene 75/25 copolymer showing the first and second heating plus the cooling curves
Conclusions
From the present and previous results, we can conclude that the Et(Ind)2ZrCl2-MAO initiator system studied is capable of homopolymerizing styrene and, to a smaller extent, copolymerizing it with cyclohexene and with 1-methyl-1-cyclohexene, while norbornene turned out to be more reactive than both cyclohexene and 1-methyl-1-cyclohexene. The PS and the resulting copolymers are amorphous, with lower Tg values than PS, suggesting a less rigid material. Comparatively, styrene/(1-octadecene) copolymerization under similar reaction conditions was more efficient, producing S/(1-C18H36) block copolymer as the proportion of octadecene in the initial feed increased.
The results obtained on styrene/cycloalkene copolymerization confirm our previous findings regarding the critical influence of the monomer’s steric hindrance on its incorporation in the growing polymer chain.
References
Po R, Cardi N (1996) Synthesis of syndiotactic polystyrene: reaction mecha-nism and catalysis. Prog Polym Sci 21:47–88
Schellenberg J, Tomotsu N (2002) Syndiotactic polystyrene catalysts and polymerization. Prog Polym Sci 27:1925–1982
Dragutan V, Streck R (eds) (2000) Studies in Surface Science and Catalysis 131: Catalytic polymerization of cycloolefins Ionic, Ziegler-Natta, and Ring-Opening Metathesis Polymerization. Elsevier, Science B.V., Amsterdam
Holohan Jr. JF, Penn JY, Vredenburgh WA (1980) In: Kirk RE, Othmer DF (eds) Encyclopedia of Chemical Technology, vol 12, 3rd edn. Wiley, New York, pp 852–869
Kaminsky W (2004) The discovery of metallocene catalysts and their present state of the art. J Polym Sci Part A: Polym Chem 42(16):3911–3921
Kaminsky W, Bark A, Dake A (1990) In: Kei T, Soga K (eds) Catalytic Olefin Polymerization. Elsevier Science Publisher, Amsterdam
Streck R (1990) In: Ymamoglu Y, Zumreoglu-Karan B, Amass AJ (eds) Olefin Metathesis and Polymerization Catalysts : Synthesis Mechanism and Utilization. Kluwer Academic Publisher, Dordrecht, pp 439–445
Streck R (1990) In: Ymamoglu Y, Zumreoglu-Karan B, Amass AJ (eds) Olefin Metathesis and Polymerization Catalysts : Synthesis Mechanism and Utilization. Kluwer Academic Publisher, Dordrecht, pp 489–515
Odian G (1981) Principles of Polymerization, 2nd edn. Wiley, New York, p 617
Boor J Jr, Youngman EA, Dimbat M (1966) Polymerization of cyclopentene, 3-methylcyclopentene, and 3-methylcyclohexene. Macromol Chem 90:26–37
Roberts WJ, Day AR (1950) A study of the polymerization of α- and β-pinene with Friedel-Crafts type catalysts. J Am Chem Soc 72:1226–1230
Tsujino T, Saegusa T, Kobayashi S, Furukawa T (1964) Polymerization of norbornene by some Ziegler-Type catalysts. Kogyo Kagaku Sasshi 67:1961–1968
Kennedy JP, Makowski HS (1967) Carbonium ion polymerization of norborne- ne and its derivatives. J Macromol Sci Chem 1:345–370
Peruch F, Cramail H, Deffieux A (1998) Homopolymerization and copolymerization of styrene with norbornene with Ni-based/MAO catalysts Macro- mol. Chem Phys 199:221–2227
Rabagliati FM, Caro CJ, Pérez MA (2002) Copolymerization of styrene by diphenylzinc-additive systems. Part III. Copolymerization of styrene/para- methylstyrene using CpTiCl3-MAO and Ph2Zn-CpTiCl3-MAO initiator systems. Bol Soc Chil Quím 47:137–144
Rabagliati FM, Pérez MA, Soto MA, Martínez de Ilarduya A, Muñoz-Guerra S (2001) Copolymerization of styrene by diphenylzinc-additive systems. I. Copolymerization of styrene/p-tert-butylstyrene by Ph2Zn-metallocene-MAO Systems. Eur Polym J 37:1001–1006
Rabagliati FM, Cancino RA, Martínez de Ilarduya A, Muñoz-Guerra S (2005) Homo- and copolymerization of styrene and 1-alkene using Ph2Zn- Et(Ind)2ZrCl2-MAO initiator systems. Eur Polym J 41:1013–1019
Cancino RA, Rodríguez FJ, Pérez MA, Rabagliati FM (2005) Styrene-alkene copolymerization by CpTiCl3-additive initiator systems. J Chil Chem Soc 50:427–430
Rabagliati FM, Muñoz LA, Quijada R (2011) Styrene copolymerization Using metallocenic initiator: homo- and copolymerization of styrene with isoprene through zirconocene-MAO initiator systems. Polym Bull 67:1425–1434
Rabagliati FM, Rodríguez FJ, Alla A, Muñoz-Guerra S (2006) Styrene/substituted styrene copolymerization using diphenylzinc-metallocene-methylaluminoxane systems. Polym Int 55:910–915
Rabagliati FM, Pérez MA, Rodríguez FJ, Caro CJ, Crispel N (2005) Further studies on styrene/styrene derivative copolymerization using combined diphenylzinc-additive initiator systems. Polym Int 54:437–441
Solomon OF, Gotesman BS (1967) Calculation of viscosity number from a single measurement. Makromol Chem 104:177–184
Macrogallery (2005) Level 4, Makin’ Polymers. Figure included with permission
Karlin KD (2010) Organometallic Chemistry, JHU Course 030.442. Dept. of Chemistry, Johns Hopkins University. Baltimore
Brookhart M, Green MLH, Wong LL (1988) Carbon-Hydrogen-Transition metal bonds. Prog Inorg Chem 36:1–124
Brookhart M, Green MLH, Parkin G (2007) Agostic interactions in transition metal compounds. Proc Nat Acad Sci, PNAS 104:6908–6914
Dougnac VN, Peoples BC, Rabagliati FM, Galland GB, Quijada R. (2012) Study on the copolymerization of propylene with norbornene using metallo-cene catalysts. Polym Bull doi: 10.1007/s00289-012-0771-5
Eisen MS. (2012) Institute of Technology. Haifa, Israel. Conference:” Heteroallylic Group 4 complexes in the polymerization of alpha-olefins: Taylor Made own Polymer”. Facultad Química y Biología, Universidad de Santiago de Chile, Santiago
Ogata K, Nishimura N, Yunokuchi T, Toyota A (2008) Copolymerization of tetracyclododecene or norbornene with styrene in presence of Ni(II) com-plex/MAO catalytic system: first example for transition metal catalyzed copo-lymerization of tetracyclododecene with styrene. J App Polym Sc 108:1562–1565
Acknowledgments
Financial support by the Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) under Project 108.5061 is gratefully acknowledged. The authors thank Laboratorio Envases, Laben-Chile (USACH) through Dr. M. J. Galotto, for DSC facilities, and Dr. F. J. Rodríguez for performing and assisting with the DSC analyses. Thanks are due to Dr. Juan del C. Guerrero, of the Facultad de Química y Biología (USACH) for running the NMR spectra.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rabagliati, F.M., Yañez, D.E., Canales, D. et al. Styrene copolymerization using a metallocene-MAO initiator system. Homo- and copolymerization of styrene with some cycloalkenes. Polym. Bull. 70, 2111–2123 (2013). https://doi.org/10.1007/s00289-013-0935-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-013-0935-y