
Abstract
PMMA–PVC polymer blend systems with LiTFSI as dopant salt were prepared by solution casting technique. Studies were then performed to explore the ionic conductivity, crystallographic structure, morphology, and thermal properties of these polymer electrolytes. XRD and SEM reveal amorphous behavior and morphologies of polymer electrolytes, respectively. Coherent length was calculated to determine the amorphousity of polymer complexes. Ionic conductivity was calculated using ac-impedance spectroscopy. DSC measurements revealed a decrease in T g, whereas T m and T d were enhanced. The thermal properties of polymer electrolytes were found to enhance upon addition of 30 wt% LiTFSI. Increase in thermal stability of polymer electrolytes were further confirmed through TGA studies.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Intense development in the field of polymer electrolytes has been the major concern for creating an alternative solid-state energy source due to the inherent advantages of these polymer electrolytes. These features include elimination of corrosive solvent and harmful gas, easy processability, suppression of lithium dendrite growth, shape configuration due to flexibility, high automated process as well as light weight [1, 2]. In general, solid polymer electrolytes (SPEs) can be widely used in various devices such as electrochromic devices, chemical sensors, solid-state reference electrode systems, supercapacitors, high vacuum electrochemistry, analog memory devices, and solar cells [2, 3]. However, the main driving force into this SPE is to explore the high performance of solid-state batteries such as secondary lithium batteries (also known as rechargeable batteries).
SPEs are of great interest in the field of solid-state batteries. Its major role is as a lithium ion charge carrier to generate ionic conductivity, leading to enhancement of energy density in the batteries through formation of thin film. In addition, it acts as an electrode separator which eliminates the requirement of an inert porous spacer between the electrolyte and electrode interface. SPEs also work as binders to insure good electrical contact with electrodes. Hence, high temperature process for conventional liquid electrolytes is eliminated as well [2].
Several aspects such as high ionic conductivity, better Li+ transference number, superior mechanical strength, excellent chemical, thermal and electrochemical stabilities are important in the investigation and research onto SPEs. The poor mechanical strength of PMMA was circumvented by introducing of PVC which acts as mechanical stiffener. Saikia et al. [4] found that PVdF-co-HFP–(PC–DEC)–LiClO4 exhibits higher ionic conductivity due to higher amorphousity. These results were confirmed by X-ray diffraction (XRD) and scanning electron microscopy (SEM) analyses. DSC measurement performed in the study also revealed that these polymer electrolytes achieve good thermal stability.
According to another study using PVdF–PVC with EC and PC as plasticizer and LiClO4 as lithium salt, XRD and SEM studies confirm the lower degree of crystallinity and formation of phase-separated morphology of polymer electrolytes, respectively. In addition, thermal stability of these polymer electrolytes was extended up to 250 °C which was preferred in the lithium battery [5]. Stephan et al. [6] suggests that the ratio of 75:25 PMMA–PVC polymer blend electrolytes doped with LiTFSI salt in the presence of PC and EC combination was operated up to 90 °C by TG–DTA analysis. Comparing with the study of Stephan et al., we also employ PMMA–PVC polymer blends containing LiTFSI, however, plasticizer is absent in this study. Plasticizer was not employed in this study because of its limitations. These shortcomings are low mechanical strength, slow evaporation, reduction in thermal, electrical and electrochemical stabilities, and narrow working voltage range as well as poor electrochemical performances. Ac-impedance study is widely used to determine the ionic conductivity of polymer electrolytes. Amorphous nature of polymer electrolytes was studied by XRD and SEM analyses, while DSC and TGA were applied to investigate thermal properties. These thermal analyses are extensively performed to investigate the properties of polymer electrolytes as a function of a change in temperature.
Experimental
Materials
PMMA with an average molecular weight of 350,000 g mol−1 (Aldrich), high molecular weight of PVC (Fluka), inorganic dopant salt, LiTFSI (Fluka) and solvent, THF (J. T. Baker) were used without further purification in this study. All the thin films of polymer electrolytes were prepared using solution casting technique.
Preparation of thin films
Prior to the preparation of the polymer electrolytes, LiTFSI was dried at 100 °C for 1 h to eliminate trace amounts of water in the material. A fixed weight ratio (70:30) of PMMA blended with PVC was employed in this study. The incorporation of LiTFSI was expressed as weight ratio. Table 1 shows the composition of polymer electrolytes and their designations. Appropriate amounts of PMMA, PVC, and LiTFSI were dissolved in THF. The solution was stirred continuously for 24 h to obtain a homogeneous mixture at room temperature. After that, the solution was cast on a glass Petri dish and allowed to evaporate slowly inside a fume hood. Eventually, a free standing thin film is produced.
Characterization
X-ray diffraction (XRD)
The amorphousity of polymer electrolytes was investigated via XRD study. The XRD patterns were recorded on a Siemens D 5000 diffractometer employed with Cu-Kα radiation (λ = 1.54060 Å) over the range of 2θ = 5°–80° at ambient temperature. Debye–Scherrer equation is used to determine the coherence length as shown below:
where λ is X-ray wavelength, θ b is glancing angle of the peak, \( \Updelta 2\theta_{\rm{b}} \) is full width at half maximum (FWHM). The peak at 2θ ≈ 16° was chosen to determine the coherence length.
Ac-impedance study
All the prepared samples were subjected to ac-impedance spectroscopy and HATR–FTIR. For impedance spectroscopy, the thicknesses of the thin films were measured using micrometer screw gauge and the ionic conductivity of the samples was determined using HIOKI 3532-50 LCR HiTESTER connected to a computer for data acquisition over a frequency range between 50 Hz and 1 MHz. The thin films were sandwiched between two stainless steel disk electrodes.
Scanning electron microscopy (SEM)
Morphology of polymer blends was studied using Leica’s SEM instrument model S440 and examined at 10 kV at room temperature. Insulator such as pure PMMA, PVC, and PMMA–PVC samples are coated with a thin layer of gold to prevent electrostatic charging on the surface.
Differential scanning calorimetry (DSC)
DSC analysis was performed via METTLER TOLEDO Thermal Analyzer that comprises of DSC 823e main unit and STARe software under nitrogen flow rate of 50 ml min−1. Approximately, 2–3 mg of samples were measured and hermetically sealed in a 40 μl aluminum crucible. First, the samples were heated sequentially from 25 to 105 °C to remove trace amount of solvent and water, and then maintained for 2 min to insure the complete evaporation. Then, the samples were cooled rapidly to 25 °C and then reheated to 450 °C at a heating rate of 10 °C min−1. The final heating scan was used to evaluate the glass transition temperature (T g), melting temperature (T m), and decomposition temperature (T d). The midpoint of endothermic reaction is assigned as T g, whereas the endothermic peak point of the melting and decomposition are denoted as T m and T d, respectively.
Thermogravimetric analysis (TGA) studies
Similarly, TGA was carried out using METTLER TOLEDO Thermal Gravimetric Analyzer which consists of TGA/SDTA851e main unit and STARe software. 2–3 mg of samples were measured and placed into 150 μl of silica crucible. The samples were then heated from 30 to 400 °C at a heating rate of 10 °C min−1 under nitrogen flow rate of 10 ml min−1.
Results
XRD and ac-impedance studies
XRD analysis has been performed and their respective diffraction patterns of PMMA–PVC, pure LiTFSI, SPE 3, SPE 6, and SPE 8 are shown in Fig. 1a–e. Sharp diffraction peaks appear at 2θ = 13.6°, 15.9°, 18.6°, 18.9°, and 21.4° revealing the crystalline behavior of pure LiTFSI. As shown in Fig. 1a, two broad peaks are observed at 2θ = 15.8° and 22.3° for PMMA–PVC polymer blends. However, these peaks are shifted to 2θ value of 17.1° and 22°, 16.9° and 22.6°, and 16° and 22.3° for SPE 3, SPE 6, and SPE 8, respectively, upon addition of LiTFSI. The coherence length of these polymer electrolytes is determined for the peak at 2θ ≈ 16° using Eq. 1. The coherence length and ionic conductivity of polymer electrolytes are reported in Fig. 2.

XRD patterns of a PMMA–PVC, b Pure LiTFSI, c SPE 3, d SPE 6, and e SPE 8

Variation of coherence length logarithm of ionic conductivity at ambient temperature with respect to different mole fractions of LiTFSI into PMMA–PVC polymer blends-based polymer electrolytes at 2θ ≈ 16 °C
SEM studies
Figure 3a–d depicts SEM images of PMMA–PVC, SPE 3, SPE 6, and SPE 8 polymer electrolytes, respectively. PMMA–PVC and SPE 3 membranes illustrate coarse texture, whereas SPE 6 and SPE 8 show smooth surface. Figure 3a and b has shown some craters with average pore size around 2 and 1 μm, respectively. On the contrary, Fig. 3c demonstrates well distribution of pores in oval shape within similar dimension, with average size of 6 μm in the polymer matrix. Some fine spherical cavities with average size of 1 μm are observed in Fig. 3d.

SEM image of a PMMA–PVC, b SPE 3, c SPE 6, and d SPE 8
DSC studies
Figure 4 illustrates DSC thermogram of PMMA–PVC, SPE 3, SPE 6, and SPE 8. The T g is shifted toward higher temperature upon incorporation of LiTFSI. T g of 125 °C is attained for SPE 3, however, the value is decreased to 118 °C when 15 wt% more of LiTFSI is added into the polymer membrane. However, T g becomes higher when 40 wt% of LiTFSI is complexed with polymer matrix.

DSC thermograms of a PMMA–PVC, b SPE 3, c SPE 6, and d SPE 8
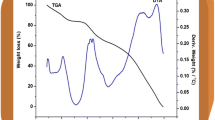
TGA studies
The TGA is reported in Fig. 5. According to Fig. 5, three apparent segments are observed for all the samples. Initially, 2–4 % of weight loss is attained at 172, 121, 139, and 130 °C for PMMA–PVC, SPE 3, SPE 6, and SPE 8, respectively. Above this initial weight loss, it remains stable up to above 252 °C for PMMA–PVC, 269 °C for SPE 3, 288 °C for SPE 6, and 279 °C for SPE 8. It is then followed up by the final mass weight loss.

TGA of PMMA–PVC, SPE 3, SPE 6, and SPE 8
Discussion
XRD and ac-impedance studies
XRD is a physical analytical method to determine the degree of crystallinity of polymer electrolytes, to prove the complexation and as well as to provide usual crystallographic data. All the sharp diffraction peaks pertaining to LiTFSI are absent, indicating the absence of uncomplexed excessive LiTFSI with no separate phase in the SPE 3 and SPE 6 polymer systems [4, 7]. Hence, it divulges that there is a complete dissolution of LiTFSI in the polymer complexes, forming complexation between PMMA–PVC with LiTFSI as well as inhibition of crystallization [8]. However, there is a sharp peak at 2θ = 13.8° for SPE 8 and this indicates the presence of excessive LiTFSI salt precipitation. These bulky ion associations obstruct the Li+ cations transportation leading to decreases in ionic conductivity as demonstrated in Fig. 2. The broad peaks at 2θ = 15.8° and 22.3° reveal the amorphous characteristic of PMMA–PVC polymer blends as portrayed in Fig. 1a. Upon inclusion of LiTFSI, the characteristic peaks exhibit some changes in peak location. This peak shifting indicates the complexation between polymer blends and lithium salt [9]. Another evidence to prove the complexation of PMMA–PVC with LiTFSI is a decrease in the peak width of characteristic peaks upon inclusion of LiTFSI. A discernible change in peak width and relative intensity of characteristic peaks for SPE 6 is illustrated in Fig. 1. These apparent peaks become broader and intensity is reduced upon addition of 30 wt% LiTFSI. These observations reveal the presence of more amorphous region in SPE 6 than SPE 3 and SPE 8 which forms a disordered arrangement in the polymer matrix. This significant disorder induces a more flexible polymer backbone thus enhancing ionic transportation. Similarly, the peaks of SPE 3 are slightly sharper than SPE 8. This suggests that SPE 8 exhibits higher degree of amorphous as sharper apparent peaks indicating higher degree of crystallinity [4].
As discussed earlier, higher amorphous degree of polymer matrix initiates the ionic hopping mechanism which contributes to higher ionic conductivity by providing a more flexible polymer chain. Therefore, the crystallite size is seminal to be determined via coherence length study. The coherence length of these polymer electrolytes is calculated for the peak at 2θ ≈ 16° in this study. As the diffraction peak width is longer, the crystallite size is shorter, contributing to lower crystallinity [10]. The increase in coherence length is in this order: SPE 6 < SPE 8 < SPE 3, as exemplified in Fig. 2. From Fig. 2, the coherence length of SPE 8 is higher than SPE 3. Therefore, it is another evidence to prove the higher amorphous degree of SPE 8 than SPE 3. In addition, this result is in good agreement with ionic conductivity at ambient temperature as illustrated in Fig. 2. The ionic conductivity of SPE 3, SPE 6, and SPE 8 are (9.12 ± 0.04) × 10−11, (1.11 ± 0.08) × 10−6, and (5.28 ± 0.06) × 10−7 S cm−1, respectively. Based on the results, the coherence length is inversely proportional to the ionic conductivity. Hence, it can be concluded that the amorphousness of a polymer matrix can take into account to improve the ionic conductivity of polymer electrolytes. From this figure, ionic conductivity of SPE 8 is higher than SPE 3. Higher amorphous nature of polymer matrix attributes to more disorder arrangement of polymeric chain and thus increases flexibility of polymer matrix. The flexible backbone could dissociate the charge carrier easily and thus raise the mobility and concentration of charge carrier. This promotes the ionic transportation within the polymer matrix and ultimately leads to higher ionic conductivity at ambient temperature. Highly flexible polymer chain has weaker interactive coordination bonds within the polymer system. Consequently, it forms more voids into the polymer system which are available for ionic migration and helps in speeding up the ionic conductivity. As discussed earlier, SPE 6 exhibits highest amorphous degree. It is further proven in coherence length measurement by showing the shortest value. It might be due to the concentration and mobility having reached the optimum level, inferring the highest ionic conductivity of SPE 6 as illustrated in Fig. 2.
SEM studies
SEM is a surface analytical characterization to provide a surface topography, crystal morphology, surface defects as well as to determine the crystallite size of polymer electrolytes [11]. The coarse texture of PMMA–PVC and SPE 3 infers higher crystallinity of polymer systems. In contrast, SPE 6 and SPE 8 polymer systems have higher amorphous nature illustrated by a smooth surface of these systems [12]. Some craters are observed in Fig. 3a, b. It is suggestive of the effect of rapid volatilization of THF solvent during preparation [13]. The decrease in these craters might be due to the interaction between LiTFSI and polymer blends. An evident change in shape of pores is observed in Fig. 3, whereby the shapes are found to have changed from rectangular to spherical with dispersion of LiTFSI. This is mainly due to the disruption of LiTFSI onto polymer blends via coordination bonding between Li+ cations with electron-rich group, such as oxygen from PMMA where the coordinating sites are C–O–C and C=O.
According to Fig. 3b, the pores with size of 9–20 μm are observed. These pores of SPE 3 are much larger than SPE 6 and SPE 8 and assigned to the coagulation of PMMA–PVC. Thus, the complexation of LiTFSI with PMMA–PVC polymer blends is lesser. Therefore, the Li+ cations migration is unfavorable and induces to lower ionic conductivity. In addition, these aggregates are surrounded by fine streak with average size of 1 μm, suggesting that PMMA–PVC polymer blends are bounded by LiTFSI salt thus allowing LiTFSI salt to start interacting with PMMA–PVC. Eventually, it forms complexation with PMMA–PVC polymer blends. The fine spherical cavities of SPE 8 are mainly due to LiTFSI salt precipitation [12]. At high LiTFSI loadings, the salts tend to exist in ion pairs rather than forming complexation with PMMA–PVC polymer blends. These aggregated ions lead to more crystalline phase in this polymer electrolyte. Eventually, these crystalline regions hinder the ionic mobility in this polymer membrane. This observation is in good agreement with XRD analysis.
The well distribution in SPE 6 infers the proper dissolution of LiTFSI and high homogeneity of polymer electrolyte with lesser agglomerates [14]. This even distribution is attributed to excellent complexation between LiTFSI and PMMA–PVC, which in turn promotes the migration of ions and eventually leads to higher ionic conductivity. In general, the main role of pores is to act as a passage for Li+ cations during charge-discharge cycle [15]. Higher porosity of SPE 6 is also exemplified in this figure, revealing the higher ions shipping.
DSC studies
DSC analysis is one of the thermal tools to elucidate thermal transition in the polymer electrolytes upon heating or cooling process under an inert atmosphere, such as nitrogen, at a controlled uniform rate [16]. The amount of heat given off by a polymer electrolyte is measured in the DSC analysis as the temperature is increased or decreased. This amount is known as heat capacity. Only one T g appears for all the samples indicating the homogeneous samples as shown in Fig. 4 [17]. T g is increased with inclusion of LiTFSI as shown in Table 1. This might be due to the formation of transient self-crosslinking as a result of coordination between Li+ cations with the electron-withdrawing groups, such as –O– and C=O functional groups [18]. This binding energy increases the barrier of the polymer matrix and reduces the degree of mobility of polymer segments. Therefore, the polymer backbone becomes less flexible [19].
As expected, T g is decreased with increasing the LiTFSI concentration. T g decreases from around 125 to 118 °C with compositions of LiTFSI from 15 to 30 wt%. This result is clearly depicted in Table 1 and Fig. 4. Plasticizing effect of LiTFSI is the main contributor. As LiTFSI loading increases, salts tend to exist in ion pairs or ion clusters between polymer matrix and thus lead to weaken the dipole–dipole interactions. As a result, the transient cross-linkage between polymer membranes is absent resulting in a decrease in solvation of Li+ cations with oxygen of ester functional group from PMMA. Eventually, a softer polymer backbone is formed resulting in enhancement of segmental motion and flexibility of polymer matrix [12]. Thus, more voids are formed as the segmental mobility is increased. Therefore, Li+ cations can easily migrate in this polymer matrix when the electric field is applied [19]. However, T g is increased with addition of 40 wt% of LiTFSI. This is suggestive of increase in formation of inter-self-crosslinking bonds between LiTFSI and polymer blends with excessive addition of LiTFSI, leading to higher barrier for rotation in the polymer chain and hence, flexibility of polymer chain becomes lesser by lowering the degree of segmental movement in the polymer chain [20].
Beyond T g, heat flow of polymer matrix becomes stable up to above 160 °C. A distinct endothermic peak is observed thereafter which is denoted as a broad melting point of electrolyte, T m. The broadening of this endothermic peak is assigned to overlapping of melting point of both PMMA and PVC. Thus, it is followed by a stable range of heat flow again over the temperature regime and then a visible sharp endothermic peak is obtained at above 300 °C. After that, a small endothermic peak is also observed as shown in Fig. 4. These peaks are corresponding to decomposition temperature of polymer electrolyte, T d. Obviously, SPE 6 exhibits highest melting temperature. This implies that the thermal properties of polymer electrolytes are enhanced with adulteration of 30 wt% of LiTFSI.
Moreover, the initial T d is associated with dehydrochlorination of PVC which is initiated by Cl free radical [21], whereas the second relatively small T d is attributed to the degradation of unsaturated group from PMMA, such as C–O bonds and degradation of PVC [17]. Overall, T d is increased upon assimilation of LiTFSI due to the interaction of LiTFSI and PMMA–PVC by forming the cross-linkage between Li+ cations and coordinating sites. Therefore, more energy is required to break the bonds and decompose the electrolytes. However, a slight decrease in T d at high LiTFSI loadings is observed. This is due to coordination of Li+ cations with oxygen or chloride. Thus, the electron density becomes lesser and eventually induces weaker unsaturated groups in the polymer matrices, such C–O bonds from PMMA [22]. As a result, SPE 6 reveals better thermal properties than SPE 3 and SPE 8 by exhibiting lowest T g and highest T m and T d.
TGA studies
TGA is another thermal technique to investigate thermal stability of polymer electrolyte under inert conditions. Weight loss of sample is monitored in an inert atmosphere over the temperature range at a controlled uniform heating rate [16]. The first small weight loss discloses the evaporation process of residue THF solvent and elimination of impurities which is mainly due to fluorine compound from LiTFSI. In addition, dehydration of moisture is also a contributor for this phenomenon as the tendency of polymer to absorb water from surrounding is quite high.
Above the first weight loss, the samples do not manifest changes in weight. Upon reaching this temperature, it starts to degrade and forms second weight loss with a dramatically mass loss. The weight loss is 59 and 35 % at 342 and 349 °C for PMMA–PVC and SPE 3, respectively. In contrast, 26 % mass loss is observed at 348 °C for SPE 6. For SPE 8, weight loss at 339 °C corresponds to 30.3 %. The second significant weight loss is mainly due to the unzipping process which contributes to a reduction in molecular weight of polymer chain at high temperature. This unzipping reaction causes many degradation reactions such as random chain scission reaction, depolymerization, intra-molecular transfer reaction whereby dimer, trimers, and oligomers are produced and inter-molecular transfer reaction as well as polymer fragments. As a result, the monomer and oligomers which chemi-adsorbed onto the polymer matrix is volatilized in this temperature regime [21]. Another attributor is the dehydrochlorination reaction from PVC, as proven in DSC thermogram.
A gradual weight loss is followed up thereafter. For PMMA–PVC and SPE 3, approximately 31 and 24 % of mass loss is noted, with residual mass of 28 and 41 % observed at 385 and 389 °C, respectively. Smaller weight losses are obtained for SPE 6 and SPE 8. SPE 6 has around 11 % weight loss at 390 °C, with residual mass of 60.35 %, whereas around 21 % of mass loss, with residual weight of 45 % at 389 °C is observed clearly for SPE 8. As a conclusion, SPE 6 is a promising candidate because its thermal stability is up to 288 °C and greatly preferred in lithium polymer batteries as its operating temperature is normally in the range of 40–70 °C [5, 6]. As aforementioned in introduction, PC–EC-based PMMA–PVC polymer blend electrolytes doped with LiTFSI is stable up to 90 °C only [6]. However, the thermal stability of our present work is greatly improved without the presence of any plasticizer. Therefore, it can be concluded that the thermal properties of free plasticizer polymer blend electrolytes is better than plasticized-based polymer blend electrolytes.
Conclusion
PMMA–PVC polymer blend electrolytes are prepared by solution casting technique. Based on analyses carried out, 70 wt% PMMA–PVC polymer blends with 30 wt% LiTFSI as dopant salt is found to be the better choice as polymer electrolyte in lithium batteries. XRD result had shown the shortest coherence length and reveals the high degree of amorphous in this polymer electrolyte. High porosity also divulges the higher degree of ionic transportation in the polymer matrix as illustrated in SEM. From DSC and TGA measurements, an excellent thermal property of the polymer matrix is observed. From DSC analysis, T g is lowered which produces a more flexible polymer backbone, whereas highest T m and T d are achieved. Thermal stability is well enhanced upon addition of 30 wt% of LiTFSI and this result is proven in TGA analysis. Higher thermal properties of PMMA–PVC–LiTFSI polymer complexes compared to plasticized-based polymer blend electrolytes make these polymer blend electrolytes are more applicable in electrochemical devices.
References
Xu JJ, Ye Hui (2005) Electrochem Commun 7:829–835
Gray FM (1991) Solid polymer electrolytes: fundamentals of technological applications. Wiley, New York, pp 1–30
Rajendran S, Sivakumar M, Subadevi R (2004) Mater Lett 58:641–649
Saikia D, Kumar A (2004) Electrochim Acta 49:2581–2589
Rajendran S, Sivakumar P (2008) Phys B 403:509–516
Stephan AM, Saito Y, Muniyandi N, Renganathan NG, Kalyanasundaram S, Elizabeth RN (2002) Solid State Ionics 148:467–473
Rajendran S, Prabhu MR, Rani MU (2008) J Power Sources 180:880–883
Sivakumar M, Subadevi R, Rajendran S, Wu HC, Wu NL (2007) Eur Polym J 43:4466–4473
Sivakumar M, Subadevi R, Rajendran S, Wu NL, Lee JY (2006) Mater Chem Phys 97:330–336
Aravindan V, Lakshmi C, Vickraman P (2009) Curr Appl Phys 9:1106–1111
Lesley S, Elaine AM (2005) Solid state chemistry: an introduction. Taylor and Francis, Boca Raton, pp 101–103
Baskaran R, Selvasekarapandian S, Kuwata N, Kawamura J, Hattori T (2007) J Phys Chem Solids 68:407–412
Stephan AM, Kumar TP, Renganathan NG, Pitchumani S, Thirunakaran R, Muniyandi N (2000) J Power Sources 89:80–87
Fonseca CG, Basaglia RMF, Brant MC, Matencio T, Domingues RZ (2009) Powder Technol 192:352–358
Uma T, Mahalingam T, Stimming U (2005) Mater Chem Phys 90:245–249
Braun D, Cherdron H, Rehahn M, Ritter H, Voit B (2005) Polymer synthesis: theory and practice. Springer, Berlin, pp 124–126
Ahmad S, Saxena TK, Ahmad S, Agnihotry SA (2006) J Power Sources 159:205–209
Li W, Yuan M, Yang M (2006) Eur Polym J. 42:1396–1402
Ramesh S, Arof AK (2001) J Power Sources 99:41–47
Yang Y, Zhou CH, Xu S, Hu H, Chen BL, Zhang J, Wu SJ, Liu W, Zhao XZ (2008) J Power Sources 185:1492–1498
Ahmad Z, Al–Awadi NA, Al–Sagheer F (2007) Polym Degrad Stab 92:1025–1033
Wu F, Feng T, Bai Y, Wu C, Ye L, Feng Z (2009) Solid State Ionics 180:677–680
Acknowledgments
This work was supported by the Exploratory Research Grant Scheme (ERGS: ER017-2011A) and Universiti Malaya Research Grant (UMRG: RG140-11AFR). One of the co-author gratefully acknowledges the “Skim Bright Sparks University Malaya (SBSUM)” for the financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ramesh, S., Liew, CW. Development and investigation on PMMA–PVC blend-based solid polymer electrolytes with LiTFSI as dopant salt. Polym. Bull. 70, 1277–1288 (2013). https://doi.org/10.1007/s00289-012-0851-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-012-0851-6