
Abstract
A new copolymer derived from 2,7-carbazole with thiophene is reported. This copolymer was obtained through the synthesis of 2,7-dibromo-N-methyl carbazole and 2-bromothiophene using a Kumada-type reaction to bond both rings and using two different kinds of Pd catalysts, [Pd(OAc)2] and [Pd(PPh3)4], with significant differences in their yields. The trimer that was obtained was subsequently electropolymerized using cyclic voltammetry, and the photoelectrochemical properties were determined. Polymerization using electrochemistry showed that the trimer contains two types of possible bonds for the formation of the macromolecule, which led to the formation of two distinctly different materials. The obtained materials exhibit promising photoelectric responses suggesting that they both can be explored in the designing of photovoltaic cells.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The growing worldwide need for electric energy produced from clean renewable sources has led to the development of new materials that help minimize the environmental impacts of current sources. One of the known alternatives for the generation of clean energy that has been thoroughly researched is photovoltaic cells [1], on which the scientific community has focused its efforts for the development of new materials with semiconducting properties that allow for a more efficient use of solar energy. In this context, conductive polymers have attracted attention in recent decades because of the possibility of the combination of their properties, such as conductivity and solar radiation absorption over a wide range of wavelengths, with the characteristics of plastics. Such combinations of properties would allow the construction of flexible, colored devices to cover large surfaces with a low processing cost [2, 3].
Among the conductive polymers, the carbazoles, also called 9-azafluorenes, are among the most interesting conjugated units for the development of polymeric films with potential electronic applications because, similar to fluorenes, they absorb a wide range of the visible spectrum. In addition, they have the advantage of being completely aromatic, electron-rich units that provide thermal and environmental stability. In addition, this base structure can be easily substituted in the 9th position, which generates a large number of structural possibilities for these materials. Moreover, carbazoles are an inexpensive starting material compared to other conjugated monomers [4]. Polymers derived from carbazole are primarily classified into two families: (i) the poly(3,6-carbazoles), which have been widely studied and are obtained directly through electrophilic substitution with halogen atoms at these positions and with subsequent carbon–carbon bonding between the units and (ii) the poly(2,7-carbazoles) [5], which are more recently synthesized units for which monomers are obtained indirectly. The poly(2,7-carbazoles) are synthesized mainly via the route proposed by Müllen and coworkers [6] (Fig. 1) with slight modifications [7, 8].

Synthesis route for of 2,7-dibromocarbazoles
The polymerization of these carbazole units is typically achieved through catalyzed carbon–carbon coupling reactions; however, the polymers obtained through this method have relatively low conductivities compared with other polymers of the same type such as polythiophene, polyanilines, and polypyrroles. For this reason, the most recent efforts in this area have been focused on obtaining copolymers that combine the properties of radiation absorption and better conductivity. This approach has been achieved by the generation of carbazole copolymers in which different structures are combined [5, 9], such as thiophenes or phenyls, through chemical polymerization. At the same time, reported theoretical results indicate that obtaining carbazole derivatives, specifically copolymers with distinct aromatic units, constitutes a good strategy for maintaining the LUMO level between 3.8 and 4.0 eV so that the band gap of the resulting polymer is between 1.2 and 1.9 eV. This strategy allows for better use of the solar spectrum [10]. With respect to conductivity, polythiophenes have high values in both their doped and neutral states, reaching values >500 S cm−1 [11].
For this reason, scientific interest has been focused on obtaining copolymers from carbazole and thiophene using previously described methods to obtain products with low molecular weight and low yields [9, 12]. Electrochemical polymerization is one of the most widely used methods for the preparation of conductive polymers [13] because of its efficiency, high degree of process control, and low cost. Recently, for the first time in the literature, the electropolymerization of units, similar to the method proposed here, has been reported as a strategy for obtaining copolymers derived from 2,7-carbazole, with thiophene, ethylenedioxythiophene, and furan as alternating units [14]. As such, the objective of this study was to synthesize the 2,7-di(thiophen-2-yl)-N-methylcarbazole (MeCzTio) trimer and perform electropolymerization with this unit. This objective is aimed at the production of a synergetic effect of the properties of the individual monomers (thiophene and 2,7-carbazole) in the polymeric material.
Experimental part
Reagents
All starting reagent and solvents were purchased from Sigma-Aldrich Co. Silica gel used for the column chromatography were purchased from Merck SA (0.063–0.200 mm and 200–400 mesh). All compounds were used without further purification.
Equipment
Polymers synthesis was performed on Radiometer Voltalab PGZ 100 model through Cyclic Voltammetry. Melting points were recorded using a Melt-Temp II equipment. FT-IR analysis was recorded using Perkin Elmer Spectrophotometer, Spectrum RX FT-IR model. NMR 1H and NMR 13C spectra were recorded on a BruckerAvance 400 MHz. Photoelectrochemical measurements were performed using a Princeton Applied Research model 273A (potentiostat/galvanostat). Illumination was performed with a Xenon Lamp of 1,000 W (Newport). Morphologic of deposits were analyzed using a ZEISS Scanning electron microscopy (SEM), model EVOMA10.
Synthesis
2,7-Dibromo-N-methylcarbazole (1)
The synthesis route employed for this compound is similar to report in literature [5] (Fig. 1).
2,7-Dibromocarbazole 1.00 g (3.08 mmol) were added to a dry 50-mL two-neck flask equipped with stir bar and condenser under N2 and DMF (15 mL) was added. NaH suspension in mineral oil 0.15 g (3.75 mmol) was added. The mixture was stirred for 30 min and iodomethane 0.4 mL (6.34 mmol) was added over 10 min. After 18 h at room temperature under N2, the reaction was quenched with water and extracted with dichloromethane. The organic layer was dried (CaCl2), filtered, an after rotary evaporation of the solvent, the light red solid is obtained. Purification on a silica column with hexane: ethyl acetate (10:1) give (1) as the white solid, needle crystals 0.79 g (2.33 mmol, 75.6 %).
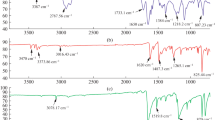
Melting point 196–198 °C. FT-IR (KBr, cm−1): 3057 w; 2925 w; 1586–1489–1451 s; 1327 m. 1H NMR (δ ppm, CDCl3): 3.78 (s, 3H, CH3);7.35 (d, J = 8.31 Hz, 2H, Cz); 7.54 (s, 2H, Cz); 7.88 (d, J = 8.31 Hz, 2H, Cz).13C NMR (δ ppm, DMSO-d 6): 29.23; 112.23; 119.04; 120.53; 121.88; 121.98; 141.64. MS (EI, 70 eV): 339 M+, 324, 260, 259, 209, 179, 151, 138, 129, 113, 98, 89, 63.
2,7-Di(thiophen-2-yl)-N-methylcarbazole
The synthesis of this compound was realized in two steps. First step is Grignard reagent preparation, for after in second step realize Kumada coupling reactions, catalyst with transition metals (Fig. 2). Two different catalysts (CH3CO2)2Pt and (P(Ph)3)4Pt were employed.

Synthesis route for monomer formation using Kumada coupling reaction
To magnesium 0.3 g (12.5 mmol) was added anhydrous THF (20 mL) under N2; 2-bromothiophene 0.9 ml (9.3 mmol) was added dropwise and the mixture was stirred at reflux for 2 h. The solution was transferred to a solution containing 2,7-dibromo-N-methylcarbazol 0.7 g (2.06 mmol) and catalyst (10 % in mol). The mixture was heated to reflux for 24 h. After addition of water, the reaction mixture was extracted with CH2Cl2 (20 mL), the organic phases were dried (MgSO4) and evaporated under reduced pressure. The resulting oil was purified by flash chromatography on silica gel (hexane/chloroform 5:1) to give pure compound as a green yellow solid. Yield was obtained using (CH3CO2)2Pt 18.3 % and (P(Ph)3)4Pt 83.0 %.
Melting point
Decomposition on 230 °C. FT-IR (KBr, cm−1): 3062 w; 2922 m; 1600–1458–1418 s; 1328 m; 690 vs; 684 vs. 1H NMR (δ ppm, CDCl3): 3.92 (s, 3H, CH3); 7.13(dd, 2H, J1 = 3.91, J2 = 0.98 Hz, thiophene); 7.31(d, 2H, J1 = 4.89, J2 = 0.98, thiophene); 7.43 (dd, 2H, J1 = 3.91, J2 = 0.98 Hz, thiophene); 7.52 (dd, 2H, J1 = 7.82, J2 = 1.47 Hz, Cz); 7.60 (s, 2H, Cz); 8.04 (d, 2H, J = 7.82 Hz, Cz).
13C NMR (δ ppm, DMSO-d 6): 29.10; 105.75; 117.20; 120.75; 121.28; 123.58; 125.30; 128.39; 131.58; 141.72; 144.50. MS (EI, 70 eV): 345 M+, 330, 300, 285, 271, 240.
Electropolymerization
Monomer electropolymerization was accomplished by either potentiodynamic sweeps (CV) or potentiostatic steps. Standard three-electrode cells were used. The working electrode (WE) was a Pt disk (0.13 cm2) or indium tin oxide ITO (1 cm2); the counter electrode was a large-area Pt wire, and the reference electrode was Ag/AgCl. All potentials in this article are reported vs Ag/AgCl. The experiments were performed at room temperature (20 °C) using as solvent anhydrous acetonitrile; electrolyte was tetrabutylammoniumhexafluorophosphate (TBAPF6). The electropolymerization of monomers was carried out from solutions containing 1 mM of monomer and 0.1 M of TBAPF6. Cyclic voltammograms were recorded with a scanning rate of 0.05 V s−1.
Photoelectrochemistry characterization
The films deposited in ITO were illuminated with a Xenon lamp of 1,000 W (Oriel Instruments 6263) arc generated with an Oriel power supply of 400–1,000 W (model 68 907). Between cell and the lamp was positioned a water filter (Oriel 61 945) to avoid heating of the solution. The photocurrent response was recorded using a potentiostat/galvanostat Applied Research Princeton (PAR) model 273-A.
Results and discussion
For the synthesis of 2,7-dibromo-N-methylcarbazole, a nitration was performed with 4,4′-dibromobiphenyl under reaction conditions that were softer than those reported in the literature [6]. In addition, a shorter chain was used in the alkylation of 2,7-dibromocarbazole, which produced a similar yield. The results obtained for the coupling reaction between this product and the Grignard reactant obtained from the Kumada coupling reaction show 83.0 % for Pd(PPh3)4 and 18.3 % for Pd(OAc)2. Based on these yield values, the best catalyst for bonding these systems is [Pd(PPh3)4]. Also with this catalyst, a smaller amount of reaction subproducts was observed through TLC, which facilitated the subsequent isolation and purification of the desired product.
Electrochemical polymerization
Figure 3 shows the potentiodynamic profile obtained during the MeCzTio electropolymerization process in the potential range between 0.3 and 1.0 V and at a scan velocity of 0.05 V s−1.

Potentiodynamic I/E profile of the methylated trimer between 0.3 and 1.0 V, with 10 potentiodynamic cycles at scan rate of 50 mVs−1, using as support electrolyte TBAPF6 0.1 M. Trimer concentration: 1 mM. Reference electrode: Ag/Ag+. WE: Pt disk. Auxiliary electrode: Pt wire
An increase was observed in the current as a function of the number of cycles, which indicates that the deposited film is conductive in nature. At least two oxidation processes that developed at anode potential values of 0.6 and 0.81 V were also observed. These processes can be attributed to the partial oxidation at particular positions of the molecule. In the return half-cycle, a wide signal was observed, which is attributed to the reduction of the previously oxidized sites. In the inset of Fig. 3, the WE can be observed with a dark colored film, following the cyclization in the middle of the study.
With the objective of separating the two observed processes, electropolymerization was performed by decreasing the anode limit to 0.75 V. Figure 4 shows the obtained results.

Potentiodynamic I/E profile of the methylated trimer between −0.3 and 0.75 V with 10 potentiodynamic cycles at scan rate of 50 mVs−1, using as support electrolyte TBAPF6 0.1 M. Trimer concentration: 1 mM. Reference electrode: Ag/Ag+. WE: Pt disk. Auxiliary electrode: Pt wire
As in the process performed with a larger potential range (Fig. 3), an increase in current is observed as a function of the number of cycles, which indicates the deposition of a conducting film on the surface of the electrode. In addition, only one diffusion-limited oxidation process is observed at 0.62 V, which may be associated with the reduction process at 0.25 V. This result indicates the occurrence of only one process, which indicates the formation of a polymeric phase that differs from the one obtained in the experiment represented in Fig. 3. Under these conditions, the polymeric film exhibits a yellow color, as observed in the inset of Fig. 4.
According to the literature, carbazole systems oxidize at potentials of ~0.7 V [15], whereas thiophene displays oxidation potentials of ~1.6 V [16]. This finding indicates that the oxidation of the carbazoles occurs at overpotentials lower than those of the thiophenes. In this context, one hypothesis is that the oxidation peak located at lower overpotentials (see Fig. 3) represents the oxidation of the carbazole ring, whereas the one that appears at the higher overpotentials is associated with the thiophenes bonded at positions 2 and 7. As such, the resulting polymer is composed of the bonding between the carbazole rings at positions 3 and 6, in addition to the carbon bonds of the thiophenes. The physical characteristics with respect to the color of the deposits support this hypothesis because, according to the literature, polycarbazole deposits are typically yellow, whereas thiophene deposits are dark colored. In addition, an electrochromic effect is observed in all the assays performed by cyclic voltammetry, i.e., yellow-green coloration in the reduced states and golden-black tones in the oxidized state.
Electrochemical polymer characterization
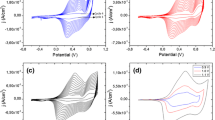
The redox properties of two deposits were investigated via cyclic voltammetry to determine how polymer oxidation/reduction potentials, corresponding to HOMO/LUMO energy levels. The used method is based on the detailed quantum chemical studies of Bredas et al. [17]; HOMO energy is given from oxidation potential and LUMO energy from reduction potential against the Ag/Ag+ reference electrode. From the cyclic voltammetry results shown in Fig. 5, the potentiodynamic profile shows peak for oxidation process at 0.59 V and reduction process at −1.32 V to deposit obtained at 0.75 V. In the case of deposit obtained at 1.1 V the values are 0.59 and −1.6 V for oxidation–reduction process. In both cases, the current of the oxidation is higher that the reduction, indicating that the two polymer are good p-type materials and poor n-type materials. The calculated band gap from onset potentials is again found to be 1.9 and 2.2 eV. Values and potentiodynamic profile prove that these two deposits have different structures, and both can be applied in the manufacture of solar cells.

Potentiodynamic I/E profile of the polymers at scan rate of 100 mVs−1, using as support electrolyte TBAPF6 0.1 M. Reference electrode: Ag/Ag+. WE: polymer deposit in Pt disk. Auxiliary electrode: Pt wire. a Deposit obtained at 0.75 V, scan between −1.8 and 0.75 V; b deposit obtained at 1.1 V, scan between −1.8 and 1.1 V
SEM analyses
Figure 6 shows SEM micrographs of the ITO substrate and polymeric phase obtained through chronoamperometry at a 0.7 V potential during 60 s. This potential was determined from the oxidation peaks observed in cyclic voltammetry, while such a time was required to cover the electrode with the polymeric surface.

SEM micrographs of the polymeric deposit generated using chronoamperometry at 0.7 V on ITO by chronoamperometry during 60 s. using as support electrolyte TBAPF6 0.1 M. Trimer concentration: 1 mM. Reference electrode: Ag/Ag+. Auxiliary electrode: Pt wire. a Bare ITO, 10,000×. Polymer: b 100×; c 10,000×; and d 40,000×
As evident in Fig. 6, the deposit is homogeneous, with agglomerations with spherical morphologies. The approximate size of the formed aggregates is 250 nm. In addition, a high degree of coverage of the surface of the electrode of ITO is evident. In Fig. 7, SEM micrographs of deposits obtained through chronoamperometry at a potential of 0.98 V are shown.

SEM micrographs of the polymeric deposit synthesized at 0.98 V on ITO by chronoamperometry during 60 s. using as support electrolyte TBAPF6 0.1 M. Trimer concentration: 1 mM. Reference electrode: Ag/Ag+. Auxiliary electrode: Pt wire. a 100×; b 1,000×; c 2,500×; d 10,000×; e and f 20,000×
Figure 7 shows that the deposits obtained at 0.98 V also display a high degree of coverage. However, this polymer exhibits a cauliflower-shaped aggregate morphology that is more heterogeneous than the one observed in the case of the polymer synthesized at 0.7 V. This potential was determined from the oxidation peaks observed in cyclic voltammetry, while such a time was required to cover the electrode with the polymeric surface.
According to the obtained results, the morphology of the deposits depends on the synthesis potential. This result supports our hypothesis that the value of the potential applied on the surface activates different positions of the used trimer; this activation induces distinct growth, which influences the final morphology.
Photocurrent measurements
Figure 8 shows the photoelectrochemical response of the films synthesized at 0.98 and at 0.7 V (Fig. 6a and b, respectively) in a medium of 0.5 M K2SO4 under polychromatic illumination.

Potentiodynamic I/E profiles of film MeCzTio deposited by chronoamperometry on ITO a at 0.98 V and b at 0.7 V during 60 s. The black line indicates deposition under darkness conditions, the red line indicates deposition under continuous illumination, and the blue line represents deposition under intermittent illumination
In general terms, a significant electrochemical signal is not observed in the absence of illumination for either polymeric phase. The observed currents can be attributed to capacitive currents that result from the ordering of the electric double layer. Under illumination, both phases display a photoresponse that is associated with a Faradaic effect that occurs at the polymer/electrolyte interface. This process has been attributed to a hydrogen evolution reaction (HER). This HER is possibly a product of the photogeneration of electrons in the LUMO of the polymer, which can be transferred to the electrolytic medium due to the potential applied to the interface. In comparative terms, the HER is initiated on both substrates at a potential of ~0.12 V, which indicates that the processes on both electrodes are of the same nature thermodynamically. However, significant differences are observed in the current obtained on the electrodes, which indicates differences in the speed of the reaction produced on these substrates. For the film deposited at 0.98 V (Fig. 7a), in the initial states and at the same potential value (i.e., 0.0 V), the resulting current for the substrate is greater than the one obtained at 0.7 V. This result confirms the hypothesis that the polymer deposited at 0.98 V has a higher conductivity than one deposited at 0.7 V, which is attributed to the bonds in the polymeric chain through the thiophene rings. Finally, when the system is intermittently illuminated at potential values that are approximately more negative than −0.6 V, an increase in the current is observed with respect to the continuously illuminated system. This result is attributed to the fact that, at this potential value, the polymer has a curvature in the conduction band that allows for more efficient charge-carrier separation and, with this curvature, a reduction in its recombination. Consequently, the resulting current increases slightly.
Conclusions
This study shows that the synthesis of the 2,7-dithiophenyl-N-methylcarbazole trimer with good yields is possible using [Pd(PPh3)4] as a catalyst. In addition, we have demonstrated that, during the electropolymerization process of these units, two oxidation processes occur that can be attributed to bonds at the 3, 6 positions of the carbazole ring and another at the 2, 5 positions of the thiophene ring. When a lower anodic overpotential is applied, electropolymerization may occur through the carbazole rings, which generates a polymer with lower conductivity and a more homogeneous morphology. In turn, the formation of the polymer at a higher overpotential would occur both through the carbazole rings and through the thiophene rings. The band-gap values for the deposits indicate that these are two different polymer structures and can be used in solar dispositives. According to the electrochemical behavior under illumination, both polymers exhibit a significant photocurrent; however, they are not free from recombination events. In subsequent studies, we will focus on a more complete photoelectrochemical characterization with the goal of determining the velocity constants of the Faradaic processes and the velocity constants of the recombination that occurs on the surface of these polymers, thereby establishing the potential applications of these materials in the production of organic photovoltaic devices.
References
Goetzbergera A, Heblinga C, Schock HW (2003) Photovoltaic materials, history, status and outlook. Mater Sci Eng R Rep 40:1–46
Günes S, Neugebauer H, Sariciftci NS (2007) Conjugated polymer-based organic solar cells. Chem Rev 107:1324–1338
Cheng YJ, Yang SH, Hsu CS (2009) Synthesis of conjugated polymers for solar cell applications. Chem Rev 109:5868–5923
Morin JF, Leclerc M, Adès D, Siove A (2005) Polycarbazoles: 25 years of progress. Macromol Rapid Commun 26:761–778
Boudreault PT, Beaupré S, Leclerc M (2010) Polycarbazoles for plastic electronics. Polym Chem 1:127–136
Dierschke F, Grimsdale AC, Müllen K (2003) Efficient SYNTHESIS OF 2,7-DIBROMOCARBAZOLES AS COMPONENTS FOR ELECTROACTIVE MATERIALS. Synthesis 16:2470–2472
Appukkuttan P, Van der Eycken E, Dehaen W (2005) Microwave-enhanced Cadogan cyclization: an easy access to the 2-substituted carbazoles and other fused heterocyclic systems. Synlett 01:127–133
Freeman W, Urvoy M, Criswell ME (2005) Triphenylphosphine-mediated reductive cyclization of 2-nitrobiphenyls: a practical and convenient synthesis of carbazoles. J Org Chem 70:5014–5019
Bouchard J, Wakim S, Leclerc M (2004) Synthesis of diindolocarbazoles by Cadogan reaction: route to ladder oligo(p-aniline)s. J Org Chem 69:5705–5711
Blouin N, Michaud A, Gendron D, Wakim S, Bair E, Neagu-Plesu R, Belletête M, Durocher G, Tao Y, Leclerc M (2008) Toward a rational design of poly(2,7-carbazole) derivatives for solar cells. J Am Chem Soc 130:732–742
Sato M, Tanaka S, Kaeriyama KJ (1985) Electrochemical preparation of highly conducting polythiophene films. J Chem Soc Chem Commun 11:713–714
Zotti G, Schiavon G, Zechin S, Morin JF, Leclerc M (2002) Electrochemical, conductive, and magnetic properties of 2,7-carbazole-based conjugated polymers. Macromolecules 35:2122–2128
Heinze J, Fontana-Uribe BA, Ludwings S (2010) Electrochemistry of conducting polymers, persistent models and new concepts. Chem Rev 110:4724–4771
Kawabata K, Goto H (2010) Electrosynthesis of 2,7-linked polycarbazole derivatives to realize low-bandgap electroactive polymer. Synth Met 160:2290–2298
Wei Z, Xu J, Nie G, Du Y, Pu S (2006) Low-potential electrochemical polymerization of carbazole and its alkyl derivatives. J Electroanal Chem 589:112–119
Roncali J (1992) Conjugated poly(thiophenes): synthesis, functionalization, and applications. Chem Rev 92:711–738
Bredas JL, Silbey R, Boudreaux DS, Chance RR (1983) Chain-length dependence of electronic and electrochemical properties of conjugated systems: polyacetylene, polyphenylene, polythiophene, and polypyrrole. J Am Chem Soc 105:6555–6559
Acknowledgments
The authors acknowledge financial support from FONDECYT Project No. 11080073. Acknowledges DEA/PUCV and CONICYT for doctorate scholarship for financial support of J.A.A., L.B., J.C.A.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Aristizabal, J.A., Soto, J.P., Ballesteros, L. et al. Synthesis, electropolymerization, and photoelectrochemical characterization of 2,7-di(thiophen-2-yl)-N-methylcarbazole. Polym. Bull. 70, 35–46 (2013). https://doi.org/10.1007/s00289-012-0817-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-012-0817-8