
Abstract
Polyelectrolyte complexation, as one simple and promising method for preparing nanoparticles, is employed to find the use in the delivery of protein drugs. Using this method, we fabricated one kind of novel nanoparticles based on two natural polysaccharides, which are the negatively charged carboxymethyl pachyman (CMP) and the positively charged chitosan (CS). The major effect factors on the average particle size, polydispersity, and zeta potential of the nanoparticles were studied. The research indicated that the physicochemical properties of the nanoparticles were deeply affected by the molecular weight, concentration, and the ionic content of two polysaccharides. The mean particle size of CMP/CS nanoparticles was almost in the range of 100–200 nm for various preparation conditions. The morphology of nanoparticles characterized by a transmission electron microscope was spherical in shape with smooth surface structure. In order to study the feasibility of these nanoparticles as oral protein delivery carriers, the encapsulation efficiency of CMP/CS nanoparticles for bovine serum albumin (BSA) was evaluated for optimized condition. It turned out that the encapsulation efficiency of BSA-loaded CMP/CS nanoparticles varied from 30.1 to 52.9% depending on the initial loading concentration of BSA as well as the concentration of CMP and CS employed in particle formation, which indicated that the concentration of polymers and drugs were all contributed to the encapsulation efficiency of nanoparticles. This report opened up another interesting perspective to develop these natural polysaccharides with emerging new applications, which have great potentials in application in the nanoparticulate delivery system.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nowadays, with the advances in biotechnology and biochemistry and the discovery of many bioactive molecules based on peptides and proteins, much effort has been devoted to developing protein delivery systems. Unfortunately, parenteral administration of proteins is still the major problem in protein drugs [1]. The challenging task in the development of protein pharmaceuticals is to deal with physical and chemical instabilities of proteins [2]. However, many research show that proteins have poor absorption efficiency in patients when administered through the parenteral route. Therefore, a large amount of work has focused on protein delivery by oral route [3, 4]. In order to achieve the successful oral delivery of protein drugs, the most important thing is to protect protein drugs from the harsh environment of gastric media in the stomach before they were absorbed in the intestine. For the last 10 years, many strategies have been developed to enhance oral protein and vaccine delivery efficiencies [5–8]. Among all these promising approaches, nanoparticulate systems have attracted especial interests. Because they can protect drugs from degradation [9], reduce toxic side effects, provide controlled release properties for encapsulated drugs due to the biodegradability, pH, and/or temperature sensibility of nanoparticle materials [10], improve drug transmucosal transport [11] and transcytosis by M cells [12, 13], and so forth. As a novel drug delivery system, nanoparticles show great potentials in biological, medical, and pharmaceutical fields [14].
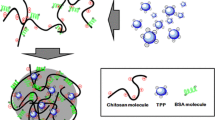
Among a wide variety of techniques available to produce nanoparticles, polyelectrolyte complexation (PEC) has received increasing attention, for the reason that the nanoparticles prepared by this method have several characteristics favorable for cellular uptake and colloidal stability, including suitable diameter and surface charge, spherical morphology and a low polydispersity index (PdI), and so on [15]. What’s more, this method is the best choice to avoid many kinds of aggression in harsh conditions, such as organic solvents and sonication during preparation. Also, this method can carry out in completely aqueous condition and in ambient temperature which do not adversely affect the stability and biological activity of these therapeutic agents [16, 17]. Figure 1 is the schematic illustration of the nanoparticles formed by polyelectrolyte complexation. Recent studies have shown that PEC is a potentially useful technique used to fabricate protein drug delivery systems for peroral administration [18].

The schematic illustration of the nanoparticles formed by polyelectrolyte complexation
Among various polymeric materials, polysaccharides are the most popular ones to prepare nanoparticles for drug delivery [19, 20]. Chitosan is a biodegradable cationic polysaccharide that is widely used for pharmaceutical applications [21]. It is the deacetylated form of chitin, which is the second abundant polysaccharide in nature [22]. Chitosan possesses many favorable biological properties of polymeric carriers [23], such as low toxicity and high susceptibility to biodegradation [24, 25], mucoadhesive properties [26], and an important capability to enhance drug permeability and absorption at mucosal sites [27, 28]. More importantly, with an estimated intrinsic pKa value of 6.5, from a technological point of view, CS is also one kind of interesting materials, for it is polycationic in acidic media and can interact with negatively charged species to form the CS complexes and nanoparticulate structures by means of ionic gelation or polyelectrolyte complexes [29, 30].
Pachyman, as a fungus polysaccharide, is a naturally occurring linear polysaccharide produced by a sclerotium of Poria cocos [31, 32], which is one of the most important herbs in China and many other Asian countries. It is composed by 1,3-β-linked d-glucose units (Fig. 2), which is in similar structure character to starch and cellulose. Up to now, pachyman is confirmed to have many pharmaceutical values, which is well known for its diuretic [33], mitogenic, complement activating [34], anti-inflammatory [35], and immunoactive properties [36]. Moreover, several derivatives of pachyman have been also prepared and developed for many pharmaceutical [37] and biomedical applications, among which the most familiar one is carboxymethyl pachyman (CMP). CMP is well known for its good water solubility and anti-tumor activity [38, 39]. By virtue of the outstanding biological and pharmacological activities, CMP has been used to fabricate drug delivery systems [40, 41].

Chemical structures of pachyman, carboxymethyl pachyman (CMP), and chitosan (CS)
According to our previous research results [42], CMP-based self-assembled nanoparticles have fabricated chemically from polymeric amphiphilic conjugates. Herein, the polyelectrolyte complexation method, which is simple and mild, was chosen to prepare nanoparticle based on CMP and CS. The processing factors affecting the characteristics of CMP/CS nanoparticles, including their physicochemical properties as well as the optimal conditions for their preparation was elucidated. Size and morphology of nanoparticles were studied. Using bovine serum albumin (BSA) as a model drug, its efficiency for protein drug association was also investigated.
Experimental
Materials
Pachyman, the average molecular weight of 2.2 × 105, extracted with 0.5 M NaOH aqueous solution from the sclerotium of Poria cocos was used [22]. Chitosan with a deacetylation degree of 95% was purchased from Jinke Biochemistry Co., Ltd. (Zhejiang, China). H2O2 was obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Bovine serum albumin (BSA) was purchased from Sigma Chemical Company (St. Louis, MO, USA). All other chemical reagents were analytical grade and obtained from commercial sources.
Synthesis of carboxymethylated pachyman (CMP)
Pachyman was carboxymethylated according to the method previously reported [42]. In brief, a suspension of 1.2 g pachyman in a mixture of 20 mL 20% NaOH and 50 mL isopropanol was stirred at an ice bath for 3 h. Then, a mixture of 10.5 g chloroacetic acid, 20 mL 20% NaOH and 50 mL isopropanol was slowly added with stirring. The reaction was continued at room temperature for 3 h and then at 50 °C for 3 h. After the solution was cooled to room temperature, 0.5 M HCl was added to adjust pH to 7.0 and then dialyzed at 4 °C by a regenerated cellulose tube (Mw cut-off 8000) against tap water for 3 days and then in distilled water for 4 days. The resulting solution was concentrated by rotary evaporator at reduced pressure below 40 °C. Finally, the carboxymethylated pachyman was lyophilized by using a lyophilizer to give the product. CMP was identified by using Fourier transform infrared spectrophotometer (FT-IR, Spectrum One, Perkin-Elmer, USA), and 1H NMR and 13C NMR (D2O) (Bruker DPX spectrometer, 400 MHz). The degree of substitution (DS) of the CMP sample was 0.9 units of carboxymethyl group per glucose determined by a method as previously described [43].
Depolymerization of chitosan
Chitosan was depolymerized with H2O2 to obtain low MW chitosan according to a method described by other literatures. [44, 45]. In brief, 5 g of chitosan were dissolved in 250 mL of 1% (v/v) acetic acid solution containing 5 g of H2O2. The solution was stirred at 25 °C for different durations, 1, 2, 3, 4, 5, 6 h, respectively. The chitosan solution was then precipitated by adding 5% (w/v) NaOH solution and adjusting pH to 7.0. The precipitates were collected by centrifugation and washed with deionized water to neutral and freeze-dried.
The intrinsic viscosity of the obtained depolymerized chitosan samples was determined at 25 °C with the co-solvent of 0.1 M acetic acid and 0.2 M sodium chloride according to the method described in other literature [46]. Calculated according to Mark–Houwink equation [46], [η] = 1.81 × 10−3 × MW0.93, the MWs of chitosan and its depolymerized samples were 45.6, 21.7, 17.0, 15.6, 15.0, 13.3, 12.6 kDa, respectively. In the following experiments, chitosan with MW of 12.6 kDa was chosen and used except for the study of MW’s effect on the complexation interaction.
Preparation of CMP/CS nanoparticles
CMP solution with different concentration (0.5, 1, 2, 3, 4, and 5 mg/mL, respectively) was prepared by dissolving the CMP powder directly into deionized water. Chitosan (CS) was dissolved in 0.25% acetic acid aqueous solution at 1 mg/mL concentration under stirring and adjusting the pH to 5.5 with 1 N NaOH. At this pH more than 90% of the amine groups of CS are protonated [17]. In the same way, chitosan solutions of different concentrations (0.5, 2, 3, 4, and 5 mg/mL) were prepared. Polyelectrolyte nanoparticles were prepared by adding 2 mL CMP solution at specified concentration (0.5, 1, 2, 3, 4, or 5 mg/mL) to 5 mL CS solution of specified concentration (0.5, 1, 2, 3, 4, or 5 mg/mL) in a beaker under gentle magnetic stirring at room temperature. Opalescent suspension was formed spontaneously and the suspension was kept under stirring for 2 h. Nanoparticles were separated from aqueous phase by ultracentrifugation (L8-80 M, Ultracentrifuge, Beckman) with 24,800 rpm at 4 °C for 40 min. The sediment was frozen at −70 °C for 5 h. Freeze drying was performed using the freeze dryer (Thermo Savant, USA).
Characterization of nanoparticles
The FT-IR spectra of the nanoparticulate samples were recorded (FT-IR, Spectrum One, Perkin-Elmer, USA) to determine the interaction between CMP and CS.
Particle size of the nanoparticles was determined by photon correlation spectroscopy using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). Samples were diluted with deionized water and the measurements were performed at a scattering angle of 90° and at temperature of 25 °C. The diameter is calculated from the autocorrelation function of the intensity of light scattered from particles, assuming a spherical form for the particles.
The polydispersity index (PdI) is a measure quantity of dispersion homogeneity which ranges from 0 to 1. Values close to 0 indicate a homogeneous dispersion while those greater than 0.3 indicate high heterogeneity [47].
The particle charge was quantified measuring the zeta potential by using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). Samples were diluted with deionized water. For the measurements samples were placed in the electrophoretic cell where a potential of 150 mV was established. The results as the mean of three determinations ± SD are presented.
The morphology of the nanoparticles was observed by using a transmission electron microscope (JEM-100CX II, Jeol Company, Japan), which is operated at an acceleration voltage of 100 kV. A drop of sample solution was placed onto a copper grid coated with carbon. After deposition for 2 min, the grid was tapped with filter paper to remove surface water, followed with air-drying.
BSA loading of nanoparticles
The drug-loaded nanoparticles were prepared by dropping a mixture of CMP and BSA into CS solution. In order to study the BSA encapsulation efficiency of the CMP/CS nanoparticles, different BSA concentration (1, 2, 3, 4, 5 mg/mL), and CMP/CS concentration ratio (1:1, 1:2, 1:3, 1:4; mg/mL: mg/mL) were used.
CMP was dissolved in 2 mL of deionized water at 1 mg/mL and different weight of BSA was added to get different BSA concentration (1, 2, 3, 4, 5 mg/mL). Then the mixed solution was dropped into a CS solution (0.25% v/v acetic acid solution) of 5 mL at a specified concentration (1, 2, 3, 4, and 5 mg/mL) under magnetic stirring at room temperature. Opalescent suspension was formed spontaneously under magnetic stirring at room temperature. The opalescent suspension was kept under stirring for 2 h. Bound and unbound BSA were separated by ultracentrifugation of the nanosuspension at 24,800 rpm at 4 °C for 40 min. The amount of free BSA in the clear supernatant was measured by UV–Vis spectrophotometry at λ = 595 nm (UV–Vis Spectrophotometer, Lambda 35, Perkin-Elmer, Norwalk, USA) using a Bradfold protein assay [48].The BSA encapsulation efficiency (EE) of the nanoparticles was calculated using the following equation.
Results and discussion
Physicochemical characterization of CMP/CS nanoparticles
Figure 3 shows the FTIR spectra of CMP, CS, and CMP-CS complex, respectively. For the IR spectrum of CMP (sodium salt form), the characteristic absorption peaks appear at 1,604 (asym., COO−) and 1,424 cm−1(sym., COO−). And in the spectrum of CS, the characteristic absorption peaks of CS are at 1,648 (amide I), 1,590 (–NH3 +), and 1,384 cm−1 (amide III). In the IR spectrum of CMP/CS, the 1,604 cm−1 peak shift to 1,590 cm−1 and the 1,424 cm−1 peak shift to 1,417 cm−1. These results indicated that the carboxylic groups of CMP had been complexed with the protonated amino groups of CS through electrostatic interactions to form the polyelectrolyte complex.

IR spectra of a CMP, b CS, and c CMP/CS nanoparticles
Effects of chitosan MW
The effects of chitosan MW on the average nanoparticle size of CMP/CS nanoparticles were investigated. In Fig. 4, the average nanoparticle size in the range of 135–279 nm enlarged as chitosan MW increased. Concerning the effects of chitosan MW on nanoparticle size, it is reasonable to believe that higher MW of chitosan leads to bigger nanoparticle size because longer chain of chitosan molecule can complex with a larger number of negatively charged CMP polymer molecules.

Effects of CS MW on the a average particle size and b zeta-potential values of CMP/CS nanoparticles ([CMP] = 1 mg/mL, [CS] = 1 mg/mL)
Commonly, the polydispersity index is an index of stability and dispersion homogeneity. All of the nanoparticles have a desired polydispersity index (0.114–0.226) less than 0.3 indicating a homogenous size distribution. But from the Fig. 4, we can see that the relationship between the MW of CS and the zeta potential of nanoparticles is not very significant.
Effect of CMP concentrations
The particle size and the size distribution of CMP/CS nanoparticles, prepared by dropwise addition of CMP into a CS solution at varying CMP concentrations, were determined and the results were displayed in Fig. 5. The data shows that the mean size of the sample increases from approximately 125–192 nm with an increase in CMP concentration. The polydispersity index of the samples in the range of 0.067–0.139 implied highly monodispersed nanoparticles. From the zeta potentials, we can see that with the increase of the CMP concentration, the potential of nanoparticles with positive charges from 50.9 to 41.4 mV decreased slightly. The effect of CMP on the zeta-potential measurement of the particles was attributed to the anionic characteristic of this polymer, which complex with CS and then reduces the surface charge of the nanoparticles.

Effects of CMP Concentrations on the a average particle sizes and b zeta-potential values of CMP/CS nanoparticles. ([CS] = 1 mg/mL)
Effects of CS concentration
The particle size and size distribution of CMP/CS nanoparticles prepared by varying the CS concentrations were determined and the results were displayed in Fig. 6. No matter that CS is prepared in 0.25% acetic acid or in 1% acetic acid solution, the mean size of the sample increases with the increase of CS solution in concentration. That phenomenon is quite reasonable, for the reason that accompanying with the increase of CS concentration, CMP molecules in one nanoparticle could interact with more chitosan molecules to make the nanoparticles become larger and larger, as shown in the Fig. 6a. However, when CS dissolved in 1% acetic acid solution, the nanoparticles formed in this solution showed slightly larger particle size under the higher CS concentration of 3–5 mg/mL than what is formed in 0.25% acetic acid solution. For the zeta potential, the nanoparticles prepared from 1% acetic acid solution displayed higher surface potential whose value was from 45.6 to 51.9 mV. The reason is that the CS were much more easier to protonate in higher acid conditions, in which the electrostatic attraction density between CS and CMP were strengthened in nanoparticles, thus resulting in increasing nanoparticle size and higher surface potential.

Effects of CS Concentrations on the a average particle sizes and b zeta-potential values of CMP/CS nanoparticles ([CMP] = 1 mg/mL)
Effects of CMP concentration and CS concentration
The particle size and size distributions of CMP/CS nanoparticles prepared by equally enhancing the CMP and CS concentration were determined and the results were displayed in Fig. 7. With the increase of the CMP and CS concentration, their nanoparticle size had a greatly increase. Nevertheless, the increase of CMP concentration and chitosan concentration is not infinite because aggregates formed easily before CMP concentration and chitosan concentration reached to a certain concentration. For example, the nanoparticles prepared from CMP or CS concentration beyond 4 mg/mL, then was prone to precipitate which is quite obvious after standing for a few days. After all, many other researchers have pointed out that the nanoparticles are prone to aggregate and precipitate if the polymer concentrations are too high. When keeping both the concentration of CMP and CS at 4 or 5 mg/mL, the polydispersity index of the nanoparticles was 0.191 or 0.228, respectively, implying that the size distribution of the samples is increasing largely. The zeta potential was increased first with the equally enhancing of CMP and CS concentration, but slight deceased for the concentration of CMP and CS at 5 mg/mL due to the aggregation and precipitation of nanoparticles (as shown in Fig. 7b).

Effects of CMP and CS concentrations on the a average particle sizes and b zeta-potential values of CMP/CS nanoparticles (X means [X] = [CMP] = [CS])
Nanoparticles morphology and distribution
The freeze-dried CMP/CS nanoparticles prepared by the PEC method were in white powdered shape and insoluble in water, dilute acidic or dilute alkali solutions. Figure 8a, b shows the TEM photograph of the nanoparticles with two different magnifications. All of the nanoparticles exhibit spherical and sub-spherical shapes with smooth surface structure.

TEM images of CMP/CS nanoparticles prepared by 1 mg/mL CMP solution and 1 mg/mL CS solution with different magnification
Figure 9a displayed the size distribution of the nanoparticles prepared by 1 mg/mL chitosan solution and 1 mg/mL CMP solution and Fig. 9b showed their zeta potential in water. The mean size and zeta potential of these CMP/CS nanoparticles were 135 nm and 47.2 mV, respectively.

a Size distribution and b zeta-potential profile of CMP/CS nanoparticles prepared by 1 mg/mL CMP solution and 1 mg/mL CS solution
BSA encapsulation efficiency (EE)
In order to study the BSA encapsulation efficiency of the CMP/CS nanoparticles, two factors, which are the BSA concentration and the CMP/CS concentration ratio were chosen and the research results were displayed in Figs. 10 and 11.

Effects of BSA concentration on the a average particle sizes, b zeta potential, and c encapsulation efficiency of BSA-loading CMP/CS nanoparticles ([CMP] = 1 mg/mL, [CS] = 1 mg/mL)

Effects of CMP/CS ratio on the a average particle sizes, b zeta-potential values, and c encapsulation efficiency of BSA-loading CMP/CS nanoparticles ([CMP] = 1 mg/mL, [BSA] = 2 mg/mL)
Notably, the BSA concentration takes an important role in the nanoparticles size and in nanoparticles’ encapsulation ability. From Fig. 10, we can see that, with the increase of the BSA concentration, the mean size of the nanoparticles increased at first, and then decreased sharply (both the CMP and CS concentration kept at 1 mg/mL). But the zeta potential of the nanoparticles almost kept decreasing with the increasing of the BSA concentration. The reason for these phenomena was related to the BSA’s physicochemical properties. It is known that the isoelectric point of BSA is in the range of 4.5–4.8 [32]. In our preparing process, the pH value of the complex system is above 5, so the negative charge of BSA was formed, which could consequently interact with the positive charged CS. But this interaction between the BSA and CS was not very obvious at first, for the reason that there was certain amounts of CMP already exist in the system. Meanwhile, the BSA encapsulation efficiency in the range of 30.1–52.9% also increased with the enhancement of the BSA concentration, which is quite reasonable when the molecular interaction between BSA and CS considered.
The polymer concentration also had great effect on the encapsulation efficiency of nanoparticles. As shown in the Fig. 11, when the CMP/CS concentration ratio kept decreasing, the average particle size increased and the encapsulation efficiency also increased first (the BSA concentration was kept at 2 mg/mL). But it is necessary to point out that, if the ratio is reduced to some extent (that is to say if the CS concentration kept increasing), the complex tends to aggregating and precipitating, which consequently weaken the ability of the nanoparticle for BSA’s encapsulation efficiency. So, when the CMP/CS ratio reached 1:4, the encapsulation efficiency of nanoparticles reduced sharply.
In one word, the encapsulation efficiency of CMP/CS nanoparticles could be optimized by adjusting the concentration of polymers and drugs. Moreover, the concentration ratio between these two opposite charged polymers also takes very important role.
Conclusions
Based on the carboxymethylated derivatives of pachyman (CMP) and CS, one novel kind of nanoparticles was prepared and characterized by polyelectrolyte complexation method. In various kinds of preparing conditions, the mean particle size of CMP/CS nanoparticles was almost in the range of 100–200 nm. Using BSA as a model drug, the encapsulation efficiency of CMP/CS nanoparticles was in the range of 30.1–52.9%, varying with the change of polymer and BSA concentrations. Drug loading research indicated that the encapsulation efficiency of the nanoparticles could be optimized by adjusting some preparation parameters, such as the initial loading concentration of BSA, an optimal concentration ratio of the concentration of CMP and CS employed for particle formation. This research is also a new way to apply this natural polysaccharide derivative, which will have great potential to be prepared as nanomaterials for protein drug delivery by a simple and moderate method.
References
Qiu B, Stefanos S, Ma J, Lalloo A, Perry BA, Leibowitz MJ, Sinko PJ, Stein S (2003) A hydrogel prepared by in situ cross-linking of a thiol-containing poly(ethylene glycol)-based copolymer: a new biomaterial for protein drug delivery. Biomaterials 24:11–18
George M, Abraham TE (2007) pH sensitive alginate–guar gum hydrogel for the controlled delivery of protein drugs. Int J Pharm 335:123–129
Ponchel G, Irache JM (1998) Specific and non-specific bioadhesive particulate systems for oral delivery to the gastrointestinal tract. Drug Delivery Rev 34:191–219
Russell-Jones GJ (2000) Oral vaccine delivery. J Controlled Release 2000(65):49–54
Wang W (1996) Oral protein drug delivery. J Drug Target 4:195–232
Kompella UB, Lee VH (2001) Delivery systems for penetration enhancement of peptide and protein drugs: design considerations. Adv Drug Deliv Rev 46:211–245
des Rieux A, Fievez V, Garinot M, Schneider YJ, Pr′eat V (2006) Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release 116:1–27
Chen F, Zhang ZR, Yuan F, Qin X, Wang M, Huang Y (2008) In vitro and in vivo study of N-trimethyl chitosan nanoparticles for oral protein delivery. Int J Pharm 349:226–233
Lowe PJ, Temple CS (1994) Calcitonin and insulin in isobutylcyanoacrylate nanocapsules: protection against proteases and effect on intestinal absorption in rats. J Pharm Pharmacol 46:547–552
Janes KA, Calvo P, Alonso MJ (2001) Polysaccharide colloidal particles as delivery systems for macromolcules. Adv Drug Del Rev 47:57–83
Gullberg E, Leonard M, Karlsson J, Hopkins AM, Brayden D, Baird AW, Artursson P (2000) Expression of specific markers and particle transport in a new human intestinal M-cell model. Biochem Biophys Res Commun 279:808–813
Galindo-Rodriguez SA, Allemann E, Fessi H, Doelker E (2005) Polymeric nanoparticles for oral delivery of drugs and vaccines: a critical evaluation of in vivo studies. Crit Rev Ther Drug Carrier Syst 22:419–464
Clark MA, Blair H, Liang L, Brey RN, Brayden D, Hirst BH (2001) Targeting polymerised liposome vaccine carriers to intestinal M cells. Vaccine 20:208–217
Zhang XG, Teng DY, Wu ZM, Wang X, Wang Z et al (2008) PEG-grafted chitosan nanoparticles as an injectable carrier for sustained protein release. J Mater Sci Mater Med 19:3525–3533
Mao H, Roy K, Troung-Le VL, Janes KA, Lin KY, Wang Y et al (2001) Chitosan-DNA nanoparticles as gene carriers: synthesis, characterization and transfection efficiency. J Control Rel 70:399–421
Silva DA, Maciel JS, Feitosa JPA, Paula HCB, de Paula RCM (2010) Polysaccharide-based nanoparticles formation by polyeletrolyte complexation of carboxymethylated cashew gum and chitosan. J Mater Sci 45:5605–5610
Mao S, Bakowsky U, Kissel T (2006) Self assembled polyelectrolyte nanocomplexes between chitosan derivatives and insulin. J Pharm Sci 95:1035–1048
Shu S, Zhang X, Teng Dg, Wang Z, Li C (2009) Polyelectrolyte nanoparticles based on water-soluble chitosan-poly(l-aspartic acid)-polyethylene glycol for controlled protein release. Carbohydr Res 344:1197–1204
Liu Z, Jiao Y, Wang Y, Zhou C, Zhang Z (2008) Polysaccharides-based nanoparticles as drug delivery systems. Adv Drug Del Rev 60:1650–1662
Krylova E, Ivanov A, Orlovski V, El-Registan G, Barinov S (2002) Hydroxypatite-polysaccharide granules for drug delivery. J Mater Sci Mater Med 13:87–90
Zhang H, Wu C, Zhang Y, White CJB, Xue Y, Nie H, Zhu L (2010) Elaboration, characterization and study of a novel affinity membrane made from electrospun hybrid chitosan/nylon-6 nanofibers for papain purification. J Mater Sci 45:2296–2304
Chen F, Zhang ZR, Huang Y (2007) Evaluation and modification of N-trimethyl chitosan chloride nanoparticles as protein carriers. Int J Pharm 336:166–173
Felt O, Buri P, Gurny R (1998) Chitosan: a unique polysaccharide for drug delivery. Drug Dev Ind Pharm 24:979–993
Onishi H, Machida Y (1999) Biodegradation and distribution of water-soluble chitosan in mice. Biomaterials 20:175–182
Lee KY, Ha WS, Park WH (1995) Blood compatibility and biodegradability of partially N-acylated chitosan derivatives. Biomaterials 16:1211–1216
Bernkop-Schurch A, Humenberg C, Valenta C (1998) Basic Studies on bioadhesive delivery systems for peptide and protein drugs. Int J Pharm 165:217–225
van der Lubben IM, Kersten G, Fretz MM, Beuvery C, Verhoef JC, Junginger HE (2003) Chitosan microparticles for mucosal vaccination against diphtheria: oral and nasal efficacy studies in mice. Vaccine 28:1400–1408
Artursson P, Lindmark T, Davis S, Illum L (1994) Effect of chitosan on the permeability of monolayers of intestinal epithelial cells (Caco-2). Pharm Res 11:1358–1361
Wang L, Wang A (2007) Adsorption characteristics of Congo Red onto the chitosan/montmorillonite nanocomposite. J Hazard Mater 147:979
Tang Y, Zhao Y, Li Y, Du Y (2010) A thermosensitive chitosan/poly(vinyl alcohol) hydrogel containing nanoparticles for drug delivery. Polym Bull 64:791–804
Chihara G, Hamuro J, Maeda Y, Arai Y, Fukuoka F (1970) Antitumour polysaccharide derived chemically from natural glucan (pachyman). Nature 225:943–944
Wang YF, Zhang M, Ruan D, Shashkov AS, Kilcoyne M, Savage AV, Zhang L (2004) Chemical components and molecular mass of six polysaccharides isolated from the sclerotium of Poria cocos. Carbohydr Res 339:327–334
Narui T, Takahashi K, Kobayashi M, Shibata S (1980) A polysaccharide produced by laboratory cultivation of Poria cocos wolf. Carbohydr Res 87:161–163
Yamada H, Kiyohara H, Takemoto N, Zhao JF, Kawamura H, Komatsu Y, Cyong JC, Aburada M, Hosoya E (1992) Studies on immunologically active substances from kampo medicine, juzen-taiho-to. 3. Mitogenic and complement activating activities of the herbal components of juzentaiho-to. Planta Med 58:166–170
Schinella GR, Tournier HA, Prieto JM, Mordujovich de Buschiazzo P, Rios JL (2002) Antioxidant activity of anti-inflammatory plant extracts. Life Sci 70:1023–1033
Wang SX, Wen YY, Hu CX (1995) Immunoactivities of the polysaccharides from Morus-alba, Chlamydomonas-mexicana and Poria-cocos. Phytother Res 9:448–451
Hu Y, Zhou X, Lu Y, Hu C, Hu X (2008) Novel biodegradable hydrogels based on pachyman and its derivatives for drug delivery. Int J Pharm 371:89–98
Hamuro J, Yamashita Y, Ohsaka Y, Maeda YY, Chihara G (1971) Carboxymethylpachymaran, a new water soluble polysaccharide with marked antitumour activity. Nature 233:486–487
Wang YF, Zhang LN, Li YQ, Hou XH, Zeng FB (2004) Correlation of structure to antitumor activities of five derivatives of a β-glucan from Poria cocos sclerotium. Carbohydr Res 339:2567–2574
Hu Y, He XR, Lei L, Liang SC, Qiu GF, Hu XM (2008) Preparation and characterization of self-assembled nanoparticles of the novel carboxymethyl pachyman-deoxycholic acid conjugates. Carbohydr Polym 74:220–227
Xiao YL, Liang SC, Qiu GF, Wu JY, Zhang JB, Hu XM (2007) Preparation, characterization and tableting properties of two new pachyman-based pharmaceutical aids: I. Disintegrants in dispersible tablets. Polym Advan Technol 18:268–274
Wang YF, Zhang LN, Ruan D (2004) Preparation and structure of five derivatives of β-(1–3)-d-glucan isolated from Poria cocos sclerotium. Chin J Polym Sci 22:137–145
Eyler RW, Klug ED, Diephuis F (1947) Determination of degree of substitution of sodium carboxymethylcellulose. Anal Chem 19:24–27
Tian F, Liu Y, Hu K, Zhao BY (2004) Study of the depolymerization behavior of chitosan by hydrogen peroxide. Carbohydr Polym 57:31–37
Liu Z, JiaoY LiuF, Zhang Z (2007) Heparin/chitosan nanoparticle carriers prepared by polyelectrolyte complexation. J Biomed Materi Res Part A 83A:806–812
Roberts GAF, Domszy JG (1982) Determination of the viscometric constants for chitosan. Int J Biol Macromol 4:374–377
Bayat A, Dorkoos FA, Dehpour AR, Moezi L, Larijani B, Junginger HE, Rafiee-Tehrani M (2008) Nanoparticles of quaternized chitosan derivatives as a carrier for colon delivery of insulin: ex vivo and in vivo studies. Int J Pharm 356:259–266
John MW (2002) The protein protocols handbook, 2nd edn. Humana Press Inc., New Jersey, p 15
Acknowledgments
This work was financially supported by the Natural Science Foundation of South-Central University for Nationalities: Studies on Nanoparticles based on Pachyman for drug delivery (YZY10009).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hu, Y., Yang, T. & Hu, X. Novel polysaccharides-based nanoparticle carriers prepared by polyelectrolyte complexation for protein drug delivery. Polym. Bull. 68, 1183–1199 (2012). https://doi.org/10.1007/s00289-011-0683-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-011-0683-9