
Abstract
Relationships able to predict the behavior of polymers based on their chemical structure are necessary for the rational synthesis of materials with desired properties. The state-of-art of polymer chemistry allows one to synthesize, using techniques such as RAFT, polymers with tailored chemical structures to study that kind of relationships. In this study, RAFT was used to synthesize copolymers—poly(methyl methacrylate)-block-poly[(2-dimethylamino)ethyl methacrylate]—that form aggregates in water at neutral and acidic pH’s. Cmc of each copolymer was determined by fluorescence using the probe N-phenyl-1-naphthylamine (NPN), and the values are consistent with the particular chemical structure of each one. These findings can be useful in future studies of dependence of cmc on the chemical structure and solution conditions and to tailor polymers with specific properties and applications, making the cmc determination faster and more accurate. To the best of our knowledge, this it the first time that NPN is used to determine the cmc of this kind of positively charged copolymers.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Relationships that would allow one to predict the aggregation properties based on the molecular structure of amphiphilic block copolymers are still under debate [1]. Although there are many attempts in that direction [2–4], robust and useful relationships relating the chemical structure to the behavior of polymers and copolymers in solution are still needed, to allow the tailoring of materials with specific properties, which would be an important improvement in efficiency of the whole process of producing systems with desired behavior. This kind of relationship is, on the other hand, well studied and defined for low molecular weight surfactants (LMWSs) [5]. Therefore, approaches to study, in a systematic way, the aggregation of different amphiphilic copolymers in solution are important to build and test reliable relationships linking the chemical structure of such materials and their solution properties, what have been already accomplished successfully for LMWS.
Amphiphilic diblock copolymers are well known to form aggregates in selective solvents, a process usually called micellization, although it seems to be different from the classic micellization of LMWS [2, 6, 7]. As in the case of LMWS, the copolymers to aggregate must have blocks with different affinities with the solvent. In water, one block must be (more) hydrophilic (“polar head”, in analogy with the LMWS) and the other one less hydrophilic (“apolar tail”).
Copolymer aggregation has been studied as resembling the micellization of LMWSs [2]. Although there are indeed several points in common, copolymer aggregation seems to be a unique phenomenon, based on the fact that the size of the aggregating molecules are usually more than 10-fold larger than usual LMWS, what leads to particular kinetics and thermodynamic parameters [8]. Therefore, a specific experimental approach to study the system is still necessary.
It is worth to emphasize that all materials classified as polymers and copolymers are a large and sometimes broad collection of molecules. They can differ by the monomers chemical structure, average total molecular weight, blocks ratio and, also, polydispersity—because nearly all polymeric materials are mixtures [9]. Hence, the study of the correlation between the aggregation properties and molecular structure of the material is a far more complex task when compared to LMWS.
Systematic change of copolymer structure and parallel determination of aggregation properties is a classic experimental approach to define empirical relationships; this has been already done in some degree, for instance, with commercially available copolymers [2, 9]. In this fashion, controlled radical polymerization (CRP) techniques provide a powerful tool to obtain copolymer materials with well-defined chemical structure and, more than that, previously designed ones. For example, CRP is an appropriate way to synthesize series of copolymers with systematic differences in their structure. If the value for a specific property (variable) is measured for each polymer in the series, it is likely to recognize the relationship connecting the changes on chemical structure to the value for that variable.
CRP techniques are one of the most growing fields in polymer science [10]. It has broken chemical limitations of free radical polymerization (FRP) allowing one to obtain, for example, amphiphilic block copolymers with low polydispersity. Among CRP variants, reversible addition-fragmentation chain transfer (RAFT) is, perhaps, the most robust, have been employed with a very large range of monomers [11] in different solvents, even in water [12]. RAFT is a convenient way to generate copolymers with specific architectures such as amphiphilic diblock copolymers.
Concerning the aggregation of block copolymers in water, one of the most important aggregation parameter is, in many ways, the critical micelle concentration (cmc) [13]. That is defined as the concentration that separate two regimes: one at low concentration where there are free molecules of the surfactant dissolved in the solvent and a second one, at high surfactant concentration, at which the molecules are forming multi-molecules aggregates called micelles. Of course, there are intermediate aggregation states in between these two regions, and the cmc depends on the method used to measure it, because the solution property followed to determine the cmc changes specifically with the surfactant concentration and the presence of pre-aggregates and micelles. Usually, cmc obtained by the same method is strongly related to the LMWS structure [5]. This critical concentration is found for micelle systems of LMWS [13] as well as for copolymers [3], being a point in common for the aggregation process of both materials.
There are many ways to measure cmc, and, as pointed out before, the cmc is dependent on how the physical property measured to determine the two regimes is affected by the aggregates. Techniques such as conductimetry [14], surface tension [15], light scattering [16], fluorescence [17], and others [18] have been widely applied. Usually, surface tension is the method of choice, although fluorescence methods are among the most accurate and suitable ones. These are based on the use of probes that have their fluorescent spectra (usually emission spectrum) changed by the properties of their vicinity (solvent). As an example, the probe N-phenyl-1-naphthylamine (NPN) has its quantum yield strongly increased when it goes from a polar medium to a less polar one. As a result, increasing the concentration of surfactant in the presence of NPN leads to a clear jump on the fluorescence intensity, easily showing when cmc is reached [19].
In this study, as a proof-of-concept, RAFT was used to generate related poly(methyl methacrylate)-block-poly[(2-dimethylethylamino)ethyl methacrylate] (PMMA-b-PDMAEMA) copolymers and their apparent cmc were determined by NPN fluorescence, in an easy and convenient way. This kind of polymer has attracted much interest because it forms aggregates in aqueous solutions that respond to pH [20], temperature [21], and ionic strength [22]. Cmc dependence on the chemical structure of these copolymers is reported here, in a step to reach constant and robust equations that predict cmc based on the copolymer structure. Although this particular system had been studied before by surface tension [23], relationships linking the copolymer chemical structure and the cmc have not been obtained yet and a more accurate method for cmc determination is lacking.
To the best of our knowledge, this it the first time that NPN is used to determine the cmc of this kind of copolymers that present positive charges at low and neutral pH’s.
Experimental
Materials
Methylmethacrylate, 99%, (MMA) was purchased from Sigma-Aldrich, and its inhibitor was removed by washing the monomer with NaOH 1% in water. DMAEMA, 98%, (Sigma-Aldrich) was used with no previous purification. Benzoyl peroxide (BPO) was obtained from Vetec Brazil and used as received. NPN, 98%, from Sigma-Aldrich, was used with no previous purification.
Copolymer synthesis
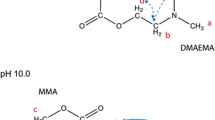
RAFT technique was used for the synthesis of PMMA-b-PDMAEMA copolymers (Fig. 1). The chain transfer agent used was cumyldithiobenzoate (CDB), previously synthesized according to a modified two-step method by Mertoglu [24], the first step being the synthesis of dithiobenzoic acid and the second one the addition of α-methylstyrene to the acid.

General chemical structure of PMMA-block-PDMAEMA synthesized by RAFT (chain transfer agent = cumyldithiobenzoate, CDB). Indexes m and n vary for each copolymer
The synthesis was started by the PMMA block, according to the parameters on Table 1. BPO was used as initiator. The PMMA blocks were purified by precipitation in methanol and drying in vacuum. These PMMA’s blocks were afterwards used as macro chain transfer agents (macroCTA’s) in a copolymerization using DMAEMA as monomer and BPO as initiator, accordingly to Table 2. The copolymers were purified by precipitation in n-hexane. Products were confirmed by 1H-NMR (INOVA DPX300 Bruker spectrometer).
All copolymers produced visually homogeneous aqueous solutions at neutral and low pH’s. Precipitation occurs within the pH range of 9–10 at 25 °C. Lower critical solution temperature (LCST) for all copolymers at pH = 7.0 is above 40 °C (data not shown).
Characterization
Gel permeation chromatography (GPC)
Gel permeation chromatography (GPC) was performed using a GPC system equipped with a Shimadzu LC20-AT HPLC solvent pump, a GSM-700 7E (Shodex) column, connected in series with differential refractometer (RID-10A, Shimadzu). The molecular weight M w was obtained via calibration curve using polystyrene standards (Polymer Labs). M w was corrected [25] according to Mark–Houwink–Sakurada equation, considering the same parameters for PMMA and the copolymer PMMA-b-PDMAEMA. THF with 0.2% of triethylamine was used as eluent at 0.6 mL min−1. All experiments were performed at room temperature.
Cmc determination by steady state fluorescence
For cmc determination fluorescent probe N-phenyl-1-naphthylamine (NPN), from Sigma–Aldrich Co., was used with no previous purification. NPN has a very low quantum yield in polar solvents as water and a higher one in less polar environments, such as copolymer aggregates. In a typical experiment, a solution of NPN in acetone was added to buffered aqueous solution to reach 2 × 10−6 mol L−1. Increasing amounts of a copolymer solution in acetone (1 × 10−3 g mL−1) were added to the previous solution under vigorous stirring, and the fluorescence signal was recorded after each addition (λexc = 340 nm, λemi = 418 nm). The plot of the ratio of fluorescence in the presence of copolymer (I) over the fluorescence in its absence (I 0, in pure aqueous buffered solution) versus the logarithm of copolymer concentration shows two regions with clear different behavior of the fluorescence ratio, one below the cmc and another one above the cmc. The cmc value was determined by the interception of the extrapolation of the logarithmic fittings for the two regions. The addition of pure acetone in aqueous solutions of NPN in the same conditions and concentrations has shown no effect on significant fluorescence. All experiments were carried out at room temperature.
Results and discussion
Figure 2 shows typical GPC traces for the materials studied. A polystyrene standard (M w = 50,000 g mol−1, PDI = 1.01) is shown in the same picture. Half-width of elution volumes are all compatible, showing the low polydispersity of the (co)polymers produced. The peak at higher elution volume is due to the PMMA macroCTA used to synthesize the copolymer 04, which is depicted in the same figure. Based on the elution volumes and compared to polystyrene standards, the average molar mass on Table 3 could be determined. In order to take into account the specific relation between hydrodynamic radius (related to elution volume) and molecular weight, correction by the Mark–Houwink–Sakurada parameters for polystyrene and polymethylmethacrylate [26] in THF was performed to calculate PMMA and PMMA-b-PDMAEMA accurate M w on Table 3. For this correction, the Mark–Houwink–Sakurada indexes were considered the same for PMMA and PMMA-b-PDMAEMA.

The cmc of each copolymer in aqueous solution is determined by experiments depicted in Fig. 3. As can be seen, NPN quantum yield changes dramatically when the aggregates form, leading the hydrophobic probe NPN to distribute between the aqueous solution (low fluorescence signal) and the aggregate less polar subphase (high fluorescence signal). The interception point from the two logarithmic fittings (below-cmc and above-cmc regimes) on each insert of Fig. 3 was considered to be the cmc for each copolymer The fluorescence signal behaves as expected, the signal-to-noise is quite good, and the two regimes are clearly indentified, proving the potential of this methodology to determine the cmc of positive-charged copolymers in aqueous solution. The results are on Table 4. For the fittings, copolymer concentrations used were up to 0.2 g L−1, because above this limit the fluorescence behavior does not follow a logarithmic trend, probably because virtually all the probe has migrated to the aggregate subphase. Cmc values are in the same range of those obtained by Chatterjee et al. for similar copolymers [23] and also by Baines et al. [27], although the total molar mass, comonomers ratio, and solution conditions presented here are different from both cases.

Fluorescence intensity ratio (I 0 = fluorescence emission at [copolymer] = 0) of NPN in aqueous solution (pH = 7.0; 5 × 10−2 mol L−1 phosphate buffer) as a function of PMMA-b-PDMAEMA copolymer concentration (C P). Copolymers are listed in Table 3. Lines are logarithmic fittings. λexc = 340 nm, λemi = 418 nm, T = 25 °C
Cmc values in Table 4 are consistent with copolymer structure parameters presented in Table 3. Those with similar structure (01–03) have cmc’s around 1 × 10−3 g L−1. Copolymer 04, which has the most dissimilar parameters (for example, the PMMA/PDMAEMA ratio and total molar mass), shows a cmc roughly 50% lower, around 0.5 × 10−3 g L−1. It is important to note that the hydrophilic/hydrophobic ratio (Table 4) of copolymers 01–03 are higher than for copolymer 04, explaining the higher cmc for the first. Such results indicate, as expected, a close relation between structure and cmc that is reproducible among polymers with comparable structures. For these kinds of copolymers this is an important finding, because the experiments were carried out in a temperature below the glass transition point of the hydrophobic block (PMMA) [27], what means that the micellar system is in the so-called “frozen” state where there is no kinetic equilibrium between unimers and micelles [8]. Even though, the concentration necessary to the unimers to assemble in such type of particle is still dependent on the structure properties such as the average molar mass and comonomer composition.
Conclusions and perspectives
It was found that RAFT is a suitable technique to synthesize PMMA-b-PDMAEMA block copolymers with systematic structure modifications to study structure and properties relationships.
NPN fluorescence is a convenient way to determine cmc of the copolymers studied, which show residual positive charge in aqueous solutions.
Concerning the cmc, the copolymers with similar structure parameters (average molar mass, hydrophilic/hydrophobic ratio, etc.) have shown very similar behavior, and the copolymer with the most distinct structure parameters has shown also the most different cmc values, compatible with its lower hydrophilic/hydrophobic ratio. Values are similar to those reported in the literature for related systems [23, 27].
The synthesis of a vast collection of similar copolymers is still necessary to reach, in the near future, valid relationships between the copolymer chemical structure and its cmc, allowing one to designed copolymers with a specific cmc.
References
Duque D (2003) Theory of copolymer micellization. J Chem Phys 119:5701–5703. doi:10.1063/1.1599279
Alexandridis P, Hatton T (2003) Poly(ethylene oxide)-poly(propylene oxide-poly(ethylene oxide) block copolymers surfactants in aqueous solutions and at interfaces: thermodynamics, structure, dynamics and modeling. Colloids Surf A 96:1–46. doi:10.1016/0927-7757(94)03028-X
Riess G (2003) Micellization of block copolymers. Prog Polym Sci 28:1107–1170. doi:10.1016/S0079-6700(03)00015-7
Zamurovic M, Christodoulou S, Vazaios A, Iatrou E, Pitsikalis M, Hadjichristidis M (2007) Micellization of complex comb like block copolymer architectures. Macromolecules 40:5835–5849. doi:10.1021/ma0704919
Blankschtein D, Thurston GM, Benedek GB (1985) Theory of phase separation in micellar solutions. Phys Rev Lett 54:955–958. doi:10.1103/PhysRevLett.54.955
Hayward RC, Pochan DJ (2010) Tailored assemblies of block copolymers in solution: it is all about the process. Macromolecules 43:3577–3584. doi:10.1021/ma9026806
Lodge TP, Muthukumar M (1996) Physical chemistry of polymers: entropy, interactions and dynamics. J Phys Chem 100:13275–13292. doi:10.1021/jp960244z
Jain S, Bates FS (2004) Consequences of nonergodicity in aqueous binary PEO-PB micellar dispersions. Macromolecules 37:1511–1523. doi:10.1021/ma035467j
Hadjichristidis N, Pispas S, Floudas GA (2003) Block copolymer. Wiley, Hoboken, NJ
Braunecker WA, Matyjaszewski K (2007) Controlled/living radical polymerization: features, developments, and perspectives. Prog Polym Sci 32:93–146. doi:10.1016/j.progpolymsci.2006.11.002
Moad G, Rizzardo E, Thang SH (2005) Living radical polymerization by the RAFT process. Aust J Chem 58:379–410. doi:10.1071/CH05072
Mccormick CL, Lowe AB (2004) Aqueous RAFT polymerization: recent developments in synthesis of functional water-soluble (Co)polymers with controlled structures. Acc Chem Res 37:312–325. doi:10.1021/ar0302484
Fendler JH (1982) Membrane mimetic chemistry. Wiley-Interscience, New York
Florenzano FH, Santos LGC, Cuccovia IM, Scarpa MV, Chaimovich H, Politi MJ (1996) Urea-induced decrease of anion selectivity in surfactant aggregates. Langmuir 12:1166–1171. doi:10.1021/la9505834
Israechivilli JN (1991) Intermolecular and surface forces. Academic Press, London
Kato T, Kanada M, Seimiya T (1995) Measurements of light scattering intensities on extremely dilute solutions of nonionic surfactant. Langmuir 11:1867–1869. doi:10.1021/la00006a009
Ananthapadmanabhan KP, Goddard ED, Turro NJ, Kuo PL (1985) Fluorescence probes for critical micelle concentration. Langmuir 1:352–355. doi:10.1021/la00063a015
Hiemenz PC (1996) Principles of colloid and surface chemistry. Marcel Dekker, New York
Zhang X, Shen Z, Feng C, Yang D, Li Y, Hu J, Lu G, Huang X (2009) PMHDO-g-PEG Double-bond-based amphiphilic graft copolymer: synthesis and diverse self-assembled nanostructures. Macromolecules 42:4249–4256. doi:10.1021/ma900343z
Fournier D, Hoogenboom R, Thijs HML, Paulus RM, Schubert US (2007) Tunable pH- and temperature-sensitive copolymer libraries by reversible addition-fragmentation chain transfer copolymerizations of methacrylates. Macromolecules 40:915–920. doi:10.1021/ma062199r
Liu R, Fraylich M, Saunders BR (2009) Thermoresponsive copolymers: from fundamental studies to applications. Colloid Polym Sci 287:627–643. doi:10.1007/s00396-009-2028-x
Ren Y, Jiang X, Yin J (2008) Copolymer of poly(4-vinylpyridine)-g-poly(ethylene oxide) respond sharply to temperature, pH and ionic strength. Eur Polym J 44:4108–4114. doi:10.1016/j.eurpolymj.2008.09.025
Chatterjee U, Jewrajka SK, Mandal BJ (2005) The amphiphilic block copolymers of 2 (dimethylamino)ethylmethacrylate and methyl methacrylate: synthesis by atom transfer radical polymerization and solution properties. Polymer 46:10699–10708. doi:10.1016/j.polymer.2005.09.045
Mertoglu M, (2004) The synthesis of well-defined functional homo- and block copolymers in aqueous media via reversible addition-fragmentation chain transfer (RAFT) polymerization. Dissertation, Postdam University
Mori S (1981) Calibration of size exclusion chromatography columns for determination of polymer molecular weight distribution. Anal Chem 53:1818–1821. doi:10.1021/ac00235a023
Bandrup J, Immergut EH, Grulke EA (1999) Polymer Handbook. Wiley, New York
Baines FL, Armes SP, Billigham NC, Tuzar Z (1996) Micellization of poly(2-(dimethylamino)ethyl methacrylate-block-methyl methacrylate) copolymers in aqueous solution. Macromolecules 29:8151–8159. doi:10.1021/ma960740l
Acknowledgments
This study was supported by Fundação de Amparo a Pesquisa de Minas Gerais (FAPEMIG), Fundação de Amparo a Pesquisa de São Paulo (FAPESP), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). VVS thanks to the Centro Universitário de Itajubá (FEPI) for support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
de Souza, V.V., Noronha, M.L.C., Almeida, F.L.A. et al. Cmc of PMMA-block-PDMAEMA measured by NPN fluorescence. Polym. Bull. 67, 875–884 (2011). https://doi.org/10.1007/s00289-011-0508-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-011-0508-x