
Abstract
A series of poly(d,l-lactide-co-glycolide) (PLGA) polymers with various molecular weight were synthesized by a ring-opening polymerization method using stannous 2-ethyl hexanoate (Sn(Oct)2) as the catalyst. The molecular weight of these polymers was controlled in a novel way, using t-butyldimethylsilanol (TBDS) or triphenylsilanol (TPS). The silicon-end group attached to the PLGA copolymer was removed at room temperature using either hydrochloric acid (HCl) or trifluoroacetic acid (TFA). The structures of these polymers before and after end group removal were characterized by 1HNMR spectroscopy, while the molecular weight and polydispersity index (PDI) were determined by viscosity method and gel permeation chromatography (GPC). The residual amounts of stannum in PLGA and the glass transition temperature (T g) of copolymer before and after end group removal were determined by the atomic absorption spectrum (AAS) and differential scanning calorimetry (DSC), respectively. The results showed that the removal method was effective. This study demonstrated that the molecular weight of PLGA could be easily controlled by altering the monomers/silanol molar ratio and the molecular weight and the purity of PLGA copolymer materials after silicon-end group removal could meet the demand of drug release.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In the research area of controlled release for drug delivery, recent focus has been on the materials used for drug coating [1–10], especially research on the drug delivery carrier prepared from biodegradable polymers. Biodegradable delivery refers to dosages of drug delivery, degradation rates, and release rates of the loaded drugs could be regulated by selecting different coating materials with the different molecular weights and compositions [11, 12].
PLGA [4–7, 13–16] exhibits excellent properties such as biocompatibility, biodegradability, and an adjustable degradation rate and hence has been widely utilized in controlled release drug delivery systems. The selection of PLGA with desired composition and molecular weight becomes critical in the research and formulation of controlled release drug delivery systems because of the important role these properties play in drug delivery applications. The composition of PLGA can be determined by controlling the feed ratio of the monomers; however, control of the molecular weight of PLGA requires extra effort.
Generally, PLGA is prepared by ring-open polymerization of lactide and glycolide using stannous 2-ethyl hexanoate (Sn(Oct)2) as the catalyst. The coordination-insertion mechanism is generally acknowledged [17, 18]: Sn(Oct)2 possesses the catalytic activity because Sn has an empty sp3d2 orbit. Trace impurities containing hydroxyl groups as the initiator (such as alcohol or water) coordinate with Sn to form the tin alkoxide. The tin alkoxide then coordinates with lactide/glycolide to push the polarization of the carbonyl group and reacts further with alcohol. Mechanism of PLGA prepared was shown in Fig. 1.

Mechanism of PLGA prepared by Sn(Oct)2 as catalyst
As described above, the hydroxyl-contained compounds play a predominant role in ring-opening polymerization of PLGA. The amount of the hydroxyl-contained compounds within a certain range affects the molecular weight of PLGA. Several types of the hydroxyl-containing compounds, such as 1-dodecanol, glycerol and 1,4-butanediol, are used for controlling the molecular weight of PLGA [19]; however, these compounds affect not only the molecular weight of the resulting polymers, but also the physical properties. In addition, the introduction of these compounds in PLGA means the introduction of the impurities, which influence the safety aspects of the PLGA used as the drug carrier in the controlled release systems could lead to unpredictable effects. Ideally, the hydroxyl-contained compounds could be removed under mild conditions after the polymerization reaction without affecting the molecular weight and the physical properties of the PLGA.
A low boiling point hydroxyl-containing compound could not be used in the ring-opening polymerization of PLGA because the reaction is carried out under high vacuum condition. We therefore chose some high boiling point hydroxyl-containing compounds to use as the initiators and to regulate the molecular weight of PLGA. In this paper, PLGA has been prepared via the ring-opening melting polymerization of d,l-lactide and glycolide, using t-butyldimethylsilanol (TBDS) or triphenylsilanol (TPS) as the molecular weight regulator and Sn(Oct)2 as a catalyst. The molecular weight of PLGA could be controlled by the silanol selection. The silicon-end group attached on the PLGA copolymer was removed at room temperature using dilute hydrochloric acid (HCl) or trifluoroacetic acid (TFA).
Experimental
Materials
Glycolide (mp 83.5–84.5 °C) and d,l-lactide (mp 126.5–127.5 °C) were purchased from Beijing Yuan-Sheng Rong Technology Co., LTD (China). t-Butyldimethylsilanol (TBDS) (bp 139.0–139.5 °C) was prepared by hydrolysis of t-butyldimethylchlorosilane (Henan Yu-Chen Fine Chemical Co., LTD, China) in our laboratory [20]. Triphenylsilanol (TPS) (mp 150–153 °C) and trimethylchlorosilane were purchased from the J&K. Trifluoroacetic acid (TFA) and pyridine were purchased from the Tianjin Chemical Reagents Sixth Factory (China). HCl was purchased from the Beijing Organic Chemical Plant (China). Sn(Oct)2 (Sigma 95%) was diluted to desired concentration (1 g/mL) with CH2Cl2. CH2Cl2 and CH3OH were distilled after treatment with anhydrous MgSO4. All chemicals were analytical grade.
Instruments
The measurements of the 1HNMR spectra were performed on a VARIAN UNITY plus 400 MHz NMR spectrometer (Varian, USA) with CDCl3 as the solvent. The intrinsic viscosity ([η]) of PLGA was determined at 25 °C with an Ubbelohde viscometer (Φ = 0.45 μm) with THF as the solvent. The corresponding viscosity-average molecular weight (M η) was calculated according to the formula [21] ([η] = 1.07 × 10−4 M 0.761η ). The weight-average molecular weight (M w) and PDI of the copolymers were determined with a Waters 510 gel permeation chromatography with HPLC grade THF as the solvent and polystyrene as the standard. Specimen concentrations were 2.5–5 mg/mL, and the flow rate was 1 mL/min at 30 °C. Differential scanning calorimetry (DSC) was performed on the NETZSCH differential scanning calorimetry with 2–3 mg sample. DSC scans were carried out over the temperature range from 0 to +80 °C at a heating rate of 5 °C/min, kept down the data. The measurements of the Atomic Absorption Spectrum (AAS) were performed on a 180-80 Polarized Zeeman Atomic Absorption Spectrophotometer (HITACHI, Japan).
Preparation of PLGA copolymer
d,l-Lactide, glycolide, the molecular weight regulator (TBDS or TPS) and Sn(Oct)2 in CH2Cl2 were mixed and kept in a silanized polymerization glass tube with a solution of trimethylchlorosilane/pyridine (15%, V/V), which was connected to a vacuum system. An exhausting–refilling process with high purity nitrogen was repeated three times and the mixture kept vacuum (20–30 Pa) about 30 min. The tube was sealed and reacted at 160 °C for 10 h. The resulting product was dissolved in CH2Cl2 and precipitated with CH3OH. The purified copolymer was dried in a vacuum oven at 40 °C for 48 h. The ratio of materials used is listed in Table 1.
Removal of silicon-end group from PLGA
A 0.50–1.00 g amount of PLGA sample, 8 mL CH2Cl2 and 8 mL HCl (or TFA) were mixed in 100 mL flask, reacted at room temperature with intense electromagnetic stirring (Table 1). The specific removal conditions used are listed in Table 3. After reaction, the product in the oil phase was separated and precipitated with CH3OH. The product was vacuum-dried at 40 °C for 48 h. Scheme of removal of the end group attached on the PLGA by acid was shown in Fig. 2.

Scheme of removal of the end group attached on the PLGA by acid
Results and discussion
The 1HNMR results of PLGA before and after removal
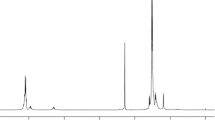
The 1HNMR of 1-PLGA-50/1 with TBDS-end group before and after removal are shown in Fig. 3a, b, respectively. As can be seen from these figures, the chemical shift at δ = 1.56, 4.82, and 5.21 ppm were assigned to the protons of Hc (–CH3), Hb (–CH2), Ha (–CH) in the PLGA, respectively [22]. Figure 3a, b presented that the chemical shift at about δ = 0.54, 1.15 ppm corresponding to the protons of He and Hd in the TBDS completely disappeared after removal reaction, which confirmed synthesis of 1-PLGA-50/1 and the TBDS-end group was successfully cleaved from the copolymer by acid.

The 1HNMR of 1-PLGA-50/1: a containing the TBDS-end group and b the TBDS-end group was removed by HCl
The 1HNMR of 2-PLGA-50/1 with TPS-end group before and after removal are shown in Fig. 4a, b, respectively. The chemical shift about at δ = 7.37–7.46, δ = 7.61–7.66 ppm corresponds to the protons of TPS. Similar results were obtained as in Fig. 3a, b, which also confirmed synthesis of 2-PLGA-50/1 and that the TPS-end group was successfully removed from copolymer.

The 1HNMR of 2-PLGA-50/1: a containing the TPS-end group and b the TPS-end group was removed by HCl
The effect of adding molecular weight regulator on the molecular weights of PLGA
Molecular weights of PLGA, as determined by viscosity methods and GPC, are shown in Table 2. Table 2 indicated that the both results were changed regularly almost at the same proportion. For 1-PLGA-50/1, the M n and M η were 7,342 and 8,537, respectively, and the PDI was 1.24. While, the M n and M η of the 1-PLGA-100/1 which prepared by less silanol were accelerated at the same time and they were 12,054 and 13,794, respectively, and the PDI was 1.23. Besides, the change trend in the percent yield of copolymer was as same as the change of PLGA M n. It shows that when the proportion of monomer/catalyst was fixed at certain reaction condition, greater silanol addition led to lower M n 1-PLGA and yield. Similar results are shown in Table 2 when the molar ratio of monomers/TPS was 50/1–400/1. When the molar ratio of monomers/TPS exceeded 400/1, the 2-PLGA M n and yield were also relatively low; because the ring-opening polymerization is a chain-polymerization process, when the ratio of monomers/silanol was too high, the active hydroxyl groups were too many preventing a high PLGA M n. When the ratio of monomers/TPS was too low, the active sites were few, making it difficult for full monomer polymerization to occur, resulting again in a low PLGA M n. The M n of PLGA prepared was 7,000–33,000 Da and PDI were rather narrow varying from 1.20 to 1.25, which could meet the requirements as a drug release materials [23].
The residual amounts of stannum in PLGA before and after end group removal
The residual amounts of stannum in PLGA before and after end group removal are shown in Table 3. It was found that the residual amounts of stannum in PLGA (1-PLGA or 2-PLGA) after end group removal were remarkably decreased compared to the amounts before removal. For the copolymer 1-PLGA-100/1, the residual amounts of stannum in PLGA before end group removal was 0.00347%, however, when the silicon-end group attached to the copolymer was removed by 2 M HCl for 24 h, the residual amounts of stannum in PLGA was decreased significantly to as low as 0.000116%; it was further observed that its value was decreased to 0.000094% when the reaction time was 27 h. It indicated that similar results were obtained from the other copolymers in Table 3. These results demonstrated that the removal methods which were in favor of the use of PLGA as carrier for drug delivery were very effective.
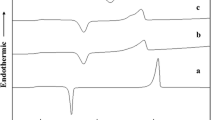
DSC analysis
The glass transition temperature (T g) of 3-PLGA, 1-PLGA-50/1, and 2-PLGA-50/1 before and after end group removal were shown in Table 4. From Table 4, it was seen that, the T g of 1-PLGA-50/1 after end group removal was 35.3 °C (M n: 6,973), which was close to the T g of 3-PLGA (35.4 °C, M n: 7,238) that added no initiator. While the T g of the 1-PLGA-50/1 before end group removal was 35.6 °C (M n: 7,342). The similar results about the 2-PLGA-50/1 were also shown in the Table 4. These results indicated to some extent that the removal after the polymerization reaction did not affect the physical properties of the PLGA.
Results of silicon-end group removal from PLGA
The silicon-end group removal from PLGA is shown in Table 5. It can be seen that the silicon-end group attached to 1-PLGA-50/1 was not completely removed using 1 M HCl for 12 h; when the reaction time was prolonged to 15 h, the end group was removed, definitively. When 2 M HCl was used, reaction time was cut to 9 h, demonstrating that the reaction could be accelerated by increasing acid concentration. This concentration and reaction time relationship was also observed with other products from Table 5. This indicated that the silicon-end group removal conditions could be mild when the PLGA M n was low, whether the polymer was 1-PLGA or 2-PLGA. Contrasted the removal result of 1-PLGA-50/1, the silicon-end group attached to 2-PLGA-50/1 was not completely removed using 1 or 2 M HCl. Only when 3 M HCl was used for 12 h, it could be removed entirely. The other copolymer also showed similar results. It could illustrate that the 1-PLGA attached by the TBDS-end group was easier removed than 2-PLGA that prepared with TPS in same conditions. There was no significant difference in the PLGA M n before and after end group removal. The molecular weight of PLGA after cleavage could meet the demands for drug-releasing materials.
Conclusions
PLGA has been prepared via the ring-opening melting polymerization of d,l-lactide and glycolide, using the molecular weight regulators TBDS and TPS and catalyst Sn(Oct)2. The molecular weight of PLGA was easily controlled by altering the monomers/silanol molar ratio. The results indicated that the molecular weight of PLGA approached to the theoretical values under all experimental conditions. The silicon-end group attached to PLGA was removed at room temperature using either HCl or TFA. 1HNMR analysis demonstrated the silicon-end group was completely cleaved from PLGA using acid of a certain concentration. The molecular weight of PLGA after cleavage was determined by GPC. Removal conditions were mild and the extent of silicon group removal could be improved by increasing the concentration of the acid and prolonging the reaction time. Besides, the residual amounts of stannum in PLGA after end group removal were much less than the amounts before removal and the T g of 1-PLGA (or 2-PLGA) after end group removal were close to the PLGA that added no initiator and had similar molecular weight. Under the same reaction conditions, the TBDS-end group attached to 1-PLGA was easier to remove than was the TPS group from 2-PLGA. The molecular weight and the purity of PLGA showed no significant difference before and after the silicon-end group was removed. The molecular weight of PLGA after cleavage could meet the requirements of drug-releasing materials.
References
Chuang CY, Don TM, Chui WY (2009) Synthesis of chitosan-based thermo- and pH-responsive porous nanoparticles by temperature-dependent self-assembly method and their application in drug release. J Polym Sci Part A: Polym Chem 47:5126–5136
Ito F, Fujimori H, Honnami H, Kawakami H, Kanamura K, Makino K (2009) Study of types and mixture ratio of organic solvent used to dissolve polymers for preparation of drug-containing PLGA microspheres. Eur Polym J 45:658–667
Zhang HH, Huang ZQ, Sun BW, Guo JX, Wang JL, Chen YQ (2008) Y-shaped poly(ethylene glycol) and poly(trimethylene carbonate) amphiphilic copolymer: synthesis and for drug delivery. J Polym Sci Part A: Polym Chem 46:8131–8140
Lee SJ, Bae Y, Kataoka K, Kim D, Lee DS, Kim SC (2008) In vitro release and in vivo anti-tumor efficacy of doxorubicin from biodegradable temperature-sensitive star-shaped PLGA-PEG block copolymer hydrogel. Polym J 40:171–176
Allison SD (2008) Effect of structural relaxation on the preparation and drug release behavior of poly(lactic-co-glycolic) acid microparticle drug delivery systems. J Pharm Sci 97:2022–2035
Sastre RL, Olmo R, Teijón C, Muñíz E, Teijón JM, Blanco MD (2007) 5-Fluorouracil plasma levels and biodegradation of subcutaneously injected drug-loaded microspheres prepared by spray-drying poly(d, l-lactide) and poly(d, l-lactide-co-glycolide) polymers. Int J Pharm 338:180–190
Matsumoto A, Matsukawa Y, Suzuki T, Yoshino H (2005) Drug release characteristics of multi-reservoir type microspheres with poly(dl-lactide-co-glycolide) and poly(dl-lactide). J Control Release 106:172–180
Liu J, Jiang ZZ, Zhang SM, Saltzman WM (2009) Poly (u-pentadecalactone-co-butylene-co-succinate) nanoparticles as biodegradable carriers for camptothecin delivery. Biomaterials 30:5707–5719
Quellec P, Gref R, Dellacherie E, Sommer F, Tran MD, Alonso MJ (1999) Protein encapsulation within poly(ethylene glycol)-coated nanospheres. II. Controlled release properties. J Biomed Mater Res 47:388–395
Han SC, He WD, Li J, Li LY, Sun XL, Zhang BY, Pan TT (2009) Reducible polyethylenimine hydrogels with disulfide crosslinkers prepared by michael addition chemistry as drug delivery carriers: synthesis, properties, and in vitro release. J Polym Sci Part A: Polym Chem 47:4074–4082
Mack BC, Wright KW, Davis ME (2009) A biodegradable filament for controlled drug delivery. J Control Release 139:205–211
Lee SJ, Park CW, Kim SC (2009) Temperature-sensitive sol-gel transition behavior of biodegradable four-arm star-shaped PEG-PLGA block copolymer aqueous solution. Polym J 41:425–431
Nahar M, Jain NK (2009) Preparation, characterization and evaluation of targeting potential of amphotericin b-loaded engineered PLGA nanoparticles. Pharm Res 26:2588–2598
Liu FJ, Guo R, Shen MW, Wang SY, Shi XY (2009) Effect of processing variables on the morphology of electrospun poly[(lactic acid)-co-(glycolic acid)] nanofibers. Macromol Mater Eng 294:666–672
Stayshich RM, Meyer TY (2008) Preparation and microstructural analysis of poly(lactic-alt-glycolic acid). J Polym Sci Part A: Polym Chem 46:4704–4711
Hamishehkar H, Emami J, Najafabadi AR, Gilani K, Minaiyan M, Mahdavi H, Nokhodchi A (2009) The effect of formulation variables on the characteristics of insulin-loaded poly(lactic-co-glycolic acid) microspheres prepared by a single phase oil in oil solvent evaporation method. Colloids Surf B 74:340–349
Zhang XC, Macdonald DA, Goosen FA, McAuley KB (1994) Mechanism of lactide polymerization in the presence of stannous octoate: the effect of hydroxy and carboxylic acid substances. J Polym Sci Part A: Polym Chem 32:2965–2970
Kricheldorf HR, Kreiser-Saunders I, Stricker A (2000) Polylactones 48. SnOct2-initiated polymerizations of lactide: a mechanistic study. Macromolecules 33:702–709
Korhonen H, Helminen A, Seppala JV (2001) Synthesis of polylactides in the presence of co-initiators with different numbers of hydroxyl groups. Polymer 42:7541–7549
Minoru T, Akira Y, Toshinobu I (1985) Production of silanol compound. JP60023385 (A)
Kenley RA, Lee MO, Mahoney TR, Sanders LM (1987) Poly(lactide-co-glycolide) decomposition kinetics in vivo and in vitro. Macromolecules 20:2398–2403
Wang ZY, Zhao YM, Wang F, Wang J (2006) Syntheses of poly(lactic acid-co-glycolic acid) serial biodegradable polymer materials via direct melt polycondensation and their characterization. J Appl Polym Sci 99:244–252
Delgado A, Evora C, Dabrés M (1998) Effect of storage on the stability of DL-PLA microspheres containing methadone. Int J Pharm 166:223–225
Acknowledgments
The authors thank the financial support of Tianjin Science and Technology Key grants (05YFGPGX26200) and the NSFC of China grants (50873114).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Ouyang, Cp., Ma, G., Zhao, Sx. et al. Preparation and characterization of the molecular weight controllable poly(lactide-co-glycolide). Polym. Bull. 67, 793–803 (2011). https://doi.org/10.1007/s00289-010-0420-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-010-0420-9