
Abstract
Wetlands are an important methane (CH4) emission source. CH4 is mainly produced during the biogeochemical process, in which methanogens and methanotrophs both play important roles. However, little is known how these two microbial communities change under different water regimes. In this study, the diversity and abundance of methanogens and methanotrophs in wetlands on Qinghai-Tibetan Plateau with different water contents (a high water content site DZ2-14-3 and a low water content site DZ2-14-4) were studied by using phylogenetic analysis and quantitative PCR based on mcrA gene and pmoA gene. A total of 16 methanogenic operational taxonomic units (OTUs) and 9 methanotrophic OTUs are obtained. For methanogens, Fen cluster (58.0%) and Methanosaetaceae (20.3%) are the dominant groups in high moisture samples, whereas Methanosaetaceae (32.4%), Methanosarcinaceae (29.4%), and Methanobacteriaceae (22.1%) are prevalent in low moisture samples. Methylobacter (90.0%) of type I methanotrophs are overwhelmingly dominant in high moisture samples, while Methylocystis (53.3%) and Methylomonas (42.2%) belonging to types II and I methanotrophs are the predominant groups in low moisture samples. Furthermore, qPCR analysis revealed that the abundance of methanogens and methanotrophs were higher in high moisture samples than that in low moisture samples. Overall, this comparative study between wetlands controlled by two different water regimes on the Qinghai-Tibetan Plateau provides fundamental data for further research on microbial functions within extreme ecosystems.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Wetlands are a large source of atmospheric CH4 and contribute 32% (~217 Tg) of global annual CH4 emissions during the decade of the 2000s [19]. Permafrost wetlands, the most fragile ecosystem threatened by climate change [55], play a vital role in terrestrial carbon storage on the Qinghai-Tibetan Plateau [20], which lies at an altitude of 4000 m above sea level (ASL) and is the largest and highest plateau on Earth [8]. A previous study has shown that the average annual CH4 emission from cold wetlands on the Qinghai-Tibetan Plateau is approximately 0.7–0.9 Tg [22], which is likely related to the abundant vegetation and the active microbial decomposition and transformation within wetlands. Therefore, microbial communities in wetland systems play an influential role in biogeochemical cycles and are crucial to the function of wetland systems [3].
Methanogens and methanotrophs are the major communities involved in CH4 cycling. The diversity and community of methanogens and methanotrophs are affected by many environmental factors, which play a significant role in governing soil microorganisms and their activities [41], such as temperature [26], soil pH [23], plant species [25], soil depth [6], land use [58], and the water regime [47]. The moisture of the soil determines the wetland type and the plant communities thriving thereon, and fluctuations in the moisture can cause changes from a methanogenic environment to CH4 oxidation environment, or vice versa [16]. Therefore, moisture is considered to have a considerable effect on CH4 emissions. Despite some investigations of the microbial communities in the wetlands have been conducted on the Qinghai-Tibetan Plateau [2, 8, 9, 17, 42, 51], differences between the methanogenic and methanotrophic microbial communities in the wetlands controlled by different water regimes have been mostly unexplored to date. Therefore, in this study, we elucidate the abundance and diversity of methanogens and methanotrophs between wetlands controlled by two different water regimes on the Qinghai-Tibetan Plateau using qPCR and phylogenetic analysis of mcrA and pmoA genes. CH4 in permafrost soils is exclusively produced by anaerobic methanogens. mcrA gene is the functional gene encoding the α subunit of methyl-coenzyme M reductase, which catalyzes the final and key enzyme in methanogenesis [46], and mcrA has been widely used for phylogenetic analysis of methanogens [35, 54]. The particulate methane monooxygenase (pMMO) gene, which encodes a key enzyme involved in CH4 oxidation [48] and Dumont and Murrell [10] found the pmoA gene to be a useful functional and phylogenetic marker for detecting methanotrophs.
Materials and Methods
Site Description and Sampling
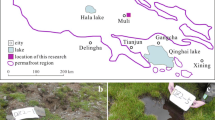
In June 2014, two sites (DZ2-14-3 and DZ2-14-4) controlled by different water regimes were selected to collect samples on the Qinghai-Tibetan Plateau (N38°04′54.04″ and E99°09′15.96″; N38°05′01.97″ and E99°09′17.12″, respectively) at an elevation of 4060 m ASL (Fig. 1). Three replicates (~5 g each) were obtained aseptically and placed into sterilized centrifuge tubes. All the samples were transported on dry ice to laboratory and stored in a −80 °C freezer until further analysis.

Two sampling sites DZ2-14-3 (a) and DZ2-14-4 (b) on the Qinghai-Tibetan Plateau
Soil Characteristics
The CH4 concentration was determined using gas chromatography (Agilent 7890A, US) and a granulometric analysis of sediments was conducted using a Mastersizer 2000 laser particle size analyzer. pH and moisture were measured as described by Wei et al. [51]. Total organic carbon (TOC) content of samples was determined according to Jiao et al. [21].
DNA Extraction and PCR Amplification
DNA was extracted using the Soil DNA Isolation Kit (MP) in accordance with the manufacturer’s instructions, and the extracted DNA was quantified by Nanodrop (ND-1000). The DNA from each replicate was mixed in equal amounts to compose the DNA of each site. mcrA and pmoA genes sequences were amplified using the primer sets mcrAF/mcrAR [34] and 189f/661r [2], respectively. PCR amplification was performed in 50 μL of mixture (including 5 µL of Taq buffer, 0.2 mM dNTPs, 1.5 mM MgCl2, 0.25 µM of each primer 5 U Taq DNA polymerase (Invitrogen, USA), and 20 ng template DNA). PCR conditions were as follows: 95 °C for 4 min followed by 30 cycles for 1 min at 94 °C, 1 min at 55 °C for mcrA gene or 52 °C (for pmoA) and 2 min at 72 °C, and a final extension step of 10 min at 72 °C. PCR products were purified using a Gel Spin DNA purification kit (AxyPrep DNA Gel Extraction Kit, USA), then ligated into the pGEM-T Easy Vector and transformed into Escherichia coli JM109 competent cells. Clones were randomly selected and sequenced.
Quantitative Polymerase Chain Reaction (qPCR)
The abundance of targeted genes were obtained using an ABI 7500 real-time PCR system (Applied Biosystems, USA). The qPCR primer pairs, ME2r/ME3mf [39] and 189f/661r [2], were used for amplifying mcrA and pmoA genes, respectively. The PCR reagent mixture contained 10 μL of SYBR Premix Ex Taq (TaKaRa, Japan), 1 μL of DNA, 10 pM of each primer, 10 pM ROX Reference Dye II, and the appropriate volume of ddH2O (with a total volume of 20 μL for qPCR). The standards for mcrA and pmoA genes were prepared from mcrA and pmoA clones that were amplified using the primer sets of mcrAF/mcrAR and 189f/661r. Plasmid was extracted and serial diluted at 1:10 to generate a standard template and they were amplified to measure the threshold cycle (CT) for a known concentration with R2 values greater than 0.99.
Phylogenetic Analyses
The sequences were determined using an ABI 3730 automated sequencer and the mcrA and pmoA genes clone libraries were constructed. Nucleotide sequences were assembled and edited using Sequencher v.4.1. OTUs were defined using a 14.3% cutoff value for mcrA and a 7% cutoff value for pmoA [2]. The coverage (C) was derived from the equation C = [1 − (n/N)] × 100, where n is the number of clones that occurred only once and N is the total number of clones examined [37]. OTUs were determined by using the computer program DOTUR [45]. Phylogenetic trees were constructed by the neighbor-joining method using the maximum-parsimony algorithm in MEGA4 software with 1000 bootstrap replicates. The functional genes mcrA and pmoA sequences were screened and translated into correct amino acid sequences for further phylogenetic analyses using DNAman software. Nucleotide sequences were then deposited in the GenBank database, and the accession numbers were as follows: mcrA clones, KX609006-KX609021 and pmoA clones, KX609022-KX609030.
Results
Characterization of Permafrost Soil
Selected soil physiochemical properties including granulometric, pH, TOC, moisture, and CH4 content of soil are shown in Table 1. Granulometric analysis reveals that throughout their stratigraphic profiles, the two samples DZ2-14-3 and DZ2-14-4 comprise sand (32.5 ± 0.2%, 9.6 ± 0.3%), silt (63.0 ± 0.3%, 77.5 ± 0.4%), and clay (4.4 ± 0.2%, 12.8 ± 0.2%). pH is slightly acidic and ranges from 6.0 ± 0.1 to 5.6 ± 0.2 between DZ2-14-3 and DZ2-14-4; The soil TOC content is 12.0 ± 0.1% and 10.2 ± 0.1% at sites DZ2-14-3 and DZ2-14-4, respectively. The moisture is also higher in site DZ2-14-3 (59.5 ± 0.3%) than that in site DZ2-14-4 (32.6 ± 0.2%); the CH4 content is 1286.7 ± 20.6 and 16.3 ± 1.2 ppm at sites DZ2-14-3 and DZ2-14-4, respectively.
Abundance of mcrA and pmoA Genes
qPCR results reveal that the mcrA and pmoA genes copies are more abundant at the high moisture site (DZ2-14-3) with 9.8 ± 0.4 × 108 copies g− 1 soil and 7.5 ± 0.4 × 107 copies g− 1 soil, respectively. In addition, the mcrA gene copies are 10 times higher at both sites than the pmoA genes.
Diversity of Methanogens Based on mcrA Gene in Both Sites
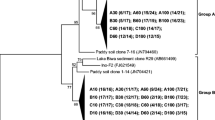
The structure of the methanogenic community revealed that 137 clones were selected and sequenced, and then assigned to 16 OTUs with coverages of 99.3% (Fig. 2). 69 sequences were obtained at the high moisture site (DZ2-14-3), Fen Cluster was the most abundant group and constituted approximately 58.0% of the clone sequences (Fig. 3). Other methanogen groups were also detected in the community phylogenetic analysis, including members of Methanosaetaceae, Rice cluster I, and Methanobacteriaceae, which comprised 20.3, 13.0, and 8.7% of the communities, respectively. 68 sequences were classified at the low moisture site (DZ2-14-4), which were clustered with the groups of Methanosaetaceae (32.4%), Methanosarcinaceae (29.4%), Methanobacteriaceae (22.1%), Fen Cluster (8.8%), and Methanomassiliicoccales (7.3%) (Fig. 3).

Phylogenetic tree showing relationships of derived amino acid sequences encoded by mcrA genes to reference sequences from the GenBank

Relative mcrA and pmoA genes abundance of methanogens and methanotrophs at the DZ2-14-3 and DZ2-14-4 sites based on gene clone libraries
Diversity of Methanotrophs Based on pmoA Gene in Both Sites
A total of 85 sequences were obtained from both sites and affiliated to 9 OTUs with coverages of 96.5% (Fig. 4). 40 sequences in the clone library were identified at the high moisture site (DZ2-14-3), and the majority of the sequences (90.0%) belonged to Methylobacter of type Ia methanotrophs, and 7.5 and 2.5% of the sequences were related to Methylomonas and Methylocystis, respectively (Fig. 3). 45 sequences were analyzed at the low moisture site (DZ2-14-4). Methylocystis (53.3%) of type II methanotrophs and Methylomonas (42.2%) of type Ia were the dominant groups, and 4.5% sequences were identified as belonging to the genus Methylococcus (Fig. 3).

Phylogenetic tree showing relationships of derived amino acid sequences encoded by pmoA genes to reference sequences from the GenBank
Discussion
Results showed evident differences between the two sites in terms of the properties of the soil and its biological characteristics. DZ2-14-3 site contained higher water content (59.5 ± 0.3 vs. 32.6 ± 0.2%), more sand (32.5 ± 0.2 vs. 9.6 ± 0.3%), and less clay (4.4 ± 0.2 vs. 12.8 ± 0.2%) relative to DZ2-14-4 site, indicating that soil at the DZ2-14-3 site is loose, multi-porous, and thus has a high capacity for water retention and a high moisture. Xiang et al. [53] indicated that a higher moisture can improve the activity of soil microorganisms. qPCR data showed that the abundance of mcrA and pmoA genes at the high moisture site (DZ2-14-3) were higher than that at the low moisture site (DZ2-14-4), which might indicate more active microbial community at the high moisture site (DZ2-14-3). The pH (6.0 ± 0.1 to 5.6 ± 0.2) of the soil was slightly acidic and similar to that measured at the other point in this area [30, 49].
However, there was a difference between the CH4 content of the soil at both sites; the high moisture site (DZ2-14-3) had a higher CH4 content than the low moisture site (DZ2-14-4). Previous research has shown that CH4 emissions increase progressively along with an increase in moisture [11, 44]. The source and sink dynamics of CH4 in soil are determined by the balance between CH4 generated by methanogens and CH4 loss caused by methanotrophic bacteria in the aerobic environment [4]. The principal influencing factor of CH4 generation is the soil’s anaerobic condition; in general, the deeper the soil layer the lower the redox potential, which provides favorable survival conditions for methanogens, leading to greater CH4 production [13]. Additionally, the DZ2-14-3 site contains more moisture compared to the DZ2-14-4 site, and has poor breathability. Accordingly, the soil environment is anaerobic, which enables extensive breeding of methanogens within the soil. In addition, as methanotroph activity is limited because of a reduction in oxygen content, the flux of CH4 emissions increases. The qPCR result shows that the gene abundance of mcrA is 10 times higher than pmoA gene. Meanwhile, Whalen et al. [52] found that CH4 diffused more easily in soil with a reduced moisture content and provided a higher CH4 oxidation rate. Despite the fact that the gene abundance of mcrA gene was higher than pmoA gene at the low moisture site, the difference was really smaller relative to that in the high moisture site (i.e., 10 times difference), which could explain that the soil moisture content might fail to meet the appropriate condition for CH4 generation.
The mcrA clone library revealed that Fen Cluster (58.0%) were the dominant groups at the high moisture site (DZ2-14-3). Fen Cluster were a family level clade within the order of Methanomicrobiales that had been widely detected where there were drastic fluctuations in the water table [54], or in acidic boreal peatlands [24] and in wetlands [31, 55]. Such results suggested that Fen Cluster methanogens were relatively tolerant to a changing water table and low pH, which agreed with previous findings determined from research on a boreal fen ecosystem [54], founding that Fen cluster characterized the dry end. Methanosaetaceae were found at the highest band percentages where acetate was the substrate [12], and Boone et al. [5] detected Methanosaetaceae utilizing only acetate. Furthermore, Peng et al. [40] suggested that Methanosaetaceae were usually predominant when the acetate concentration had already decreased to a lower steady state level of <0.2 mM. Our data showed that Methanosaetaceae occupied a higher percentage composition of methanogens (32.4%) at the low moisture site (DZ2-14-4), in accordance with the lower pH value measured at this site. Similarly, Methanobacteriaceae were distributed at both sites and frequently recovered on the Qinghai-Tibetan Plateau [8, 30, 51]; it proliferated preferentially when H2 was not limited [7]. However, Methanosarcinaceae were only detected at the low moisture site (DZ2-14-4); it existed in extensive wetlands such as peatlands, freshwater marshes [32], and paddy soils [38] due to the existence of all three known pathways for methanogenesis [59]. In freshwater wetlands, Methanosarcinaceae were able to primarily utilize acetate, various methyl compounds, and hydrogen as methanogenic substrates [5, 15]. Methanomassiliicoccus were methyl-type methanogenic archaea; they presented unique metabolic characteristics, were different from general special methyl-type methanogenic archaea, and lacked the full pathway for reducing CO2 into methyl-coenzyme M [28]. From a nutritional perspective, Methanomassiliicoccus had a mixed nutrition type [57]. Whether they played an important role in CH4 production and emission was still unclear; however, their function was attracting increasing scientific attention. Rice cluster I were originally retrieved from rice roots [14] and had recently been isolated and shown to be CO2 reducers that had a role in the inter species transfer of H2 in rice paddies [43]. Watanabe et al. [50] showed that Rice cluster I members were predominantly retrieved from soil under drained conditions. In the present study, Rice cluster I comprised 13.0% of the communities at the high moisture site (DZ2-14-3).
Analysis of the methanotrophic community revealed that Methylobacter belonging to type Ia methanotrophs were the predominant groups at the high moisture site (DZ2-14-3) (90.0%). The group had frequently been detected on the Qinghai-Tibetan Plateau [56], and Lüke et al. [33] demonstrated that the genus Methylobacter affiliated with paddy soils was indicative of high CH4 source strengths. These results were consistent with our study; the CH4 content was higher at the high moisture site (DZ2-14-3). Methylomonas, type I methanotrophs, preferred an environment with low in CH4 and high in O2 [1], and Deng et al. [8] reported Methylomonas to be detected in soils with a low moisture content. In this study, Methylomonas had the high percentage composition (42.2%) of the methanogens at the low moisture site (DZ2-14-4) but were of low relative abundance at the high moisture site (DZ2-14-3) (7.5%). In addition, Methylocystis (53.3%) were the most dominant methanotrophic groups at the low moisture site (DZ2-14-4), but only a few sequences (2.5%) were detected at the high moisture site (DZ2-14-3). Methylocystis were ubiquitous methanotrophic inhabitants of many ecosystems [27] and had often been observed on the Qinghai-Tibetan Plateau, and Leng et al. [29] reported that CH4 was the preferred substrate for Methylocystis in paddy soil. Methylocystis were more widely found in conditions with high O2 and low CH4, such as in upland agricultural soil and forestry soil [36]. Furthermore, Leng et al. [29] revealed that uncultivated Methylocystis species were facultative methanotrophs utilizing acetate as a secondary carbon source in the absence of CH4 in paddy soil. This result was in accordance with the lower CH4 content and pH value at the low moisture site (DZ2-14-4). In our study, 25 clones (29.4%) obtained from both sites had great similarities to the Methylocystis strain SB2 (ADD64440), Im et al. [18] demonstrated that strain SB2 was able to utilize not only CH4 for growth, but also ethanol and acetate.
Conclusions
In this study, we found some fundamental differences in methanogenic and methanotrophic abundance and community composition between two wetland sites under different water regimes on the Qinghai-Tibetan Plateau. qPCR analysis revealed that the abundance of methanogens and methanotrophs was higher at the high moisture site (DZ2-14-3) than that at the low moisture site (DZ2-14-4). Similarly, the CH4 content was higher at the high moisture site (DZ2-14-3). Cloning analyses of mcrA and pmoA genes revealed that Fen Cluster, Methanosaetaceae, Methylobacter were predominant at the high moisture site (DZ2-14-3), while Methanosaetaceae, Methanosarcinaceae, Methanobacteriaceae, Methylocystis, Methylomonas were the dominant groups at the low moisture site (DZ2-14-4). This comparative analysis study makes a contribution to improving scientific understanding of the microbial community structure in permafrost wetlands.
References
Arai H, Hadi A, Darung U, Limin SH, Hatano R, Inubushi K (2014) A methanotrophic community in a tropical peatland is unaffected by drainage and forest fires in a tropical peat soil. Soil Sci Plant Nutr 60:577–585
Barbier BA, Dziduch I, Liebner S, Ganzert L, Lantuit H, Pollard W, Wagner D (2012) Methane-cycling communities in a permafrost-affected soil on Herschel Island, Western Canadian Arctic: active layer profiling of mcrA and pmoA genes. FEMS Microbiol Ecol 82:287–302
Bodelier PL, Dedysh SN (2013) Microbiology of wetlands. Front Microbiol 4:1–4
Bodelier PL, Laanbroek HJ (2004) Nitrogen as a regulatory factor of methane oxidation in soils and sediments. FEMS Microbiol Ecol 47:265–277
Boone D, Castenholz R, Garrity G (2001) vol 1: the Archaea and the deeply branching and phototrophic bacteria. Springer, New York
Cadillo-Quiroz H, Bräuer S, Yashiro E, Sun C, Yavitt J, Zinder S (2006) Vertical profiles of methanogenesis and methanogens in two contrasting acidic peatlands in central New York State, USA. Environ Microbiol 8:1428–1440
Conrad R, Klose M (2006) Dynamics of the methanogenic archaeal community in anoxic rice soil upon addition of straw. Eur J of Soil Sci 57:476–484
Deng Y, Cui X, Hernández M, Dumont MG (2014) Microbial diversity in hummock and hollow soils of three wetlands on the Qinghai-Tibetan Plateau revealed by 16S rRNA pyrosequencing. Plos ONE 9:e103115
Ding W, Cai Z, Wang D (2004) Preliminary budget of methane emissions from natural wetlands in China. Atmos Environ 38:751–759
Dumont MG, Murrell JC (2005) Community-level analysis: key genes of aerobic methane oxidation. Methods Enzymol 397:413–427
Elberling B, Nordstrøm C, Grøndahl L, Søgaard H, Friborg T, Christensen TR, Ström L, Marchand F, Nijs I (2008) High-arctic soil CO2 and CH4 production controlled by temperature, water, freezing and snow. Adv Ecol Res 40:441–472
Fey A, Conrad R (2000) Effect of temperature on carbon and electron flow and on the archaeal community in methanogenic rice field soil. Appl Environ Microbiol 66:4790–4797
Fiedler S, Höll B, Jungkunst H (2005) Methane budget of a Black Forest spruce ecosystem considering soil pattern. Biogeochemistry 76:1–20
Großkopf R, Stubner S, Liesack W (1998) Novel euryarchaeotal lineages detected on rice roots and in the anoxic bulk soil of flooded rice microcosms. Appl Environ Microbiol 64:4983–4989
Hori T, Noll M, Igarashi Y, Friedrich MW, Conrad R (2007) Identification of acetate-assimilating microorganisms under methanogenic conditions in anoxic rice field soil by comparative stable isotope probing of RNA. Appl Environ Microbiol 73:101–109
Hu Q, Zhu L, Xing R, Yao B, Hu B (2011) Methane emission from a Carex-dominated wetland in Poyang Lake. Acta Ecol Sin 31:4851–4857 (In Chinese)
Huang F, Zhang Y, Zhu Y, Wang P, Lu J, Lv J (2014) Flavobacterium qiangtangensis sp. nov., isolated from Qiangtang basin in Qinghai-Tibetan Plateau, China. Curr Microbiol 69(3):234–239
Im J, Lee SW, Yoon S, DiSpirito AA, Semrau JD (2011) Characterization of a novel facultative Methylocystis species capable of growth on methane, acetate and ethanol. Environ Microbiol Rep 3:174–181
International Panel on Climate Change (IPCC) (2013) Climate change 2013: the physical science basis. Cambridge University Press, Cambridge
Jay G, Xilai L, BRIERLEY G (2012) Topographic influence on wetland distribution and change in Maduo County, Qinghai-Tibet Plateau, China. J Mount Sci 9:362–371
Jiao L, Su X, Wang Y, Jiang H, Zhang Y, Chen F (2015) Microbial diversity in the hydrate-containing and-free surface sediments in the Shenhu area, South China Sea. Geosci Front 6:627–633
Jin H, Wu J, Cheng G, Nakano T, Sun G (1999) Methane emissions from wetlands on the Qinghai-Tibet Plateau. Chin Sci Bull 44:2282–2286
Johri JK, Surange S, Nautiyal CS (1999) Occurrence of salt, pH, and temperature-tolerant, phosphate-solubilizing bacteria in alkaline soils. Curr Microbiol 39(2):89–93
Juottonen H, Tuittila E-S, Juutinen S, Fritze H, Yrjälä K (2008) Seasonality of rDNA-and rRNA-derived archaeal communities and methanogenic potential in a boreal mire. ISME J 2:1157–1168
Kao-Kniffin J, Freyre DS, Balser TC (2010) Methane dynamics across wetland plant species. Aquat Bot 93:107–113
Kim S-Y, Freeman C, Fenner N, Kang H (2012) Functional and structural responses of bacterial and methanogen communities to 3-year warming incubation in different depths of peat mire. Appl Soil Ecol 57:23–30
Kip N, van Winden JF, Pan Y, Bodrossy L, Reichart G-J, Smolders AJ, Jetten MS, Damsté JSS, den Camp HJO (2010) Global prevalence of methane oxidation by symbiotic bacteria in peat-moss ecosystems. Nat Geosci 3:617–621
Lang K, Schuldes J, Klingl A, Poehlein A, Daniel R, Brune A (2014) Comparative genome analysis of “Candidatus Methanoplasma termitum” indicates a new mode of energy metabolism in the seventh order of methanogens. Appl Environ Microbiol. https://doi.org/10.1128/AEM.03389-14
Leng L, Chang J, Geng K, Lu Y, Ma K (2014) Uncultivated Methylocystis Species in paddy soil include facultative methanotrophs that utilize acetate. Microb Ecol 70:1–9
Li Y, Su X, Zhu Y, Lu Z, Wei S, Cui H, Li L, Liu H, Zhang S (2015) Winter seasonal characteristics of the archaeal communities in different alpine ecosystem topsoils collected from the gas hydrate drilling area, Qilian mountains. Geoscience 29:1047–1060 (In Chinese)
Lin Y, Liu D, Ding W, Kang H, Freeman C, Yuan J, Xiang J (2015) Substrate sources regulate spatial variation of metabolically active methanogens from two contrasting freshwater wetlands. Appl Microbiol Biotechnol 99:10779–10791
Liu D, Ding W, Jia Z, Cai Z (2012) The impact of dissolved organic carbon on the spatial variability of methanogenic archaea communities in natural wetland ecosystems across China. Appl Microbiol Biotechnol 96:253–263
Lüke C, Krause S, Cavigiolo S, Greppi D, Lupotto E, Frenzel P (2010) Biogeography of wetland rice methanotrophs. Environ Microbiol 12:862–872
Luton PE, Wayne JM, Sharp RJ, Riley PW (2002) The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfillb. Microbiology 148:3521–3530
Martí M, Juottonen H, Robroek BJ, Yrjälä K, Danielsson Å, Lindgren P-E, Svensson BH (2015) Nitrogen and methanogen community composition within and among three Sphagnum dominated peatlands in Scandinavia. Soil Biol Biochem 81:204–211
Mei J, Wang L, Han D, Zhao Y (2011) Methanotrophic community structure of aged refuse and its capability for methane bio-oxidation. J Environ Sci 23:868–874
Mullins TD, Britschgi TB, Krest RL, Giovannoni SJ (1995) Genetic comparisons reveal the same unknown bacterial lineages in Atlantic and Pacific bacterioplankton communities. Limnol Oceanogr 40:148–158
Noll M, Klose M, Conrad R (2010) Effect of temperature change on the composition of the bacterial and archaeal community potentially involved in the turnover of acetate and propionate in methanogenic rice field soil. FEMS Microbiol Ecol 73:215–225
Nunoura T, Oida H, Miyazaki J, Miyashita A, Imachi H, Takai K (2008) Quantification of mcrA by fluorescent PCR in methanogenic and methanotrophic microbial communities. FEMS Microbiol Ecol 64:240–247
Peng J, Lü Z, Rui J, Lu Y (2008) Dynamics of the methanogenic archaeal community during plant residue decomposition in an anoxic rice field soil. Appl Environ Microbiol 74:2894–2901
Pengthamkeerati P, Motavalli P, Kremer R (2011) Soil microbial activity and functional diversity changed by compaction, poultry litter and cropping in a claypan soil. Appl Soil Ecol 48:71–80
Piao AL, Feng XM, Nogi Y, Han L, Li Y, Lv J (2016) Sphingomonas qilianensis sp. nov., isolated from surface soil in the permafrost region of Qilian Mountains, China. Curr Microbiol 72(4):363–369
Sakai S, Conrad R, Liesack W, Imachi H (2010) Methanocella arvoryzae sp. nov., a hydrogenotrophic methanogen isolated from rice field soil. Int J Syst Evol Microbiol 60:2918–2923
Schaufler G, Kitzler B, Schindlbacher A, Skiba U, Sutton M, Zechmeister-Boltenstern S (2010) Greenhouse gas emissions from European soils under different land use: effects of soil moisture and temperature. Eur J of Soil Sci 61:683–696
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Steinberg LM, Regan JM (2008) Phylogenetic comparison of the methanogenic communities from an acidic, oligotrophic fen and an anaerobic digester treating municipal wastewater sludge. Appl Environ Microbiol 74:6663–6671
Urbanová Z, Picek T, Bárta J (2011) Effect of peat re-wetting on carbon and nutrient fluxes, greenhouse gas production and diversity of methanogenic archaeal community. Ecol Eng 37:1017–1026
Vorobev AV, Baani M, Doronina NV, Brady AL, Liesack W, Dunfield PF, Dedysh SN (2011) Methyloferula stellata gen. nov., sp. nov., an acidophilic, obligately methanotrophic bacterium that possesses only a soluble methane monooxygenase. Int J Syst Evol Microbiol 61:2456–2463
Wang Y, Wei S, Cui H, Su X, Hu F, Zhu Y, Lu Z, Liu H, Zhang S, Pang S (2016) Methane metabolic microbial community structure in the active layer and the permafrost layer of the Qilian permafrost, China. Appl Environ Biol 22:592–598 (In Chinese)
Watanabe T, Cahyani VR, Murase J, Ishibashi E, Kimura M, Asakawa S (2009) Methanogenic archaeal communities developed in paddy fields in the Kojima Bay polder, estimated by denaturing gradient gel electrophoresis, real-time PCR and sequencing analyses. Soil Sci Plant Nutr 55:73–79
Wei S, Cui H, He H, Hu F, Su X, Zhu Y (2014) Diversity and distribution of archaea community along a stratigraphic permafrost profile from Qinghai-Tibetan Plateau, China. Archaea. https://doi.org/10.1155/2014/240817
Whalen SC, Reeburgh W, Sandbeck K (1990) Rapid methane oxidation in a landfill cover soil. Appl Environ Microbiol 56:3405–3411
Xiang S-R, Doyle A, Holden PA, Schimel JP (2008) Drying and rewetting effects on C and N mineralization and microbial activity in surface and subsurface California grassland soils. Soil Biol Biochem 40:2281–2289
Yrjälä K, Tuomivirta T, Juottonen H, Putkinen A, Lappi K, TUITTILA ES, Penttilä T, Minkkinen K, Laine J, Peltoniemi K (2011) CH4 production and oxidation processes in a boreal fen ecosystem after long-term water table drawdown. Global Change Biol 17:1311–1320
Yun J, Ju Y, Deng Y, Zhang H (2014) Bacterial community structure in two permafrost wetlands on the Tibetan Plateau and Sanjiang Plain, China. Microb Ecol 68:360–369
Yun J, Zhuang G, Ma A, Guo H, Wang Y, Zhang H (2012) Community structure, abundance, and activity of methanotrophs in the Zoige wetland of the Tibetan Plateau. Microb Ecol 63:835–843
Zhang J, Xu Y, Lu Y (2015) Microbial mechanisms of methane production and oxidation in terrestrial ecosystems. Acta Ecol Sin 35:6592–6603 (In Chinese)
Zheng Y, Liu X, Zhang L, Zhou Z, He J (2010) Do land utilization patterns affect methanotrophic communities in a Chinese upland red soil? J Environ Sci 22(12):1936–1943
Zinder SH (1993) Physiological ecology of methanogens Methanogenesis. Springer, New York, pp 128–206
Acknowledgements
This research was supported by the Funds of Oil and Gas Survey, China Geological Survey (No. GZH201400308).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cui, H., Su, X., Wei, S. et al. Comparative Analyses of Methanogenic and Methanotrophic Communities Between Two Different Water Regimes in Controlled Wetlands on the Qinghai-Tibetan Plateau, China. Curr Microbiol 75, 484–491 (2018). https://doi.org/10.1007/s00284-017-1407-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-017-1407-7