
Abstract
RNase E plays an important role in the degradation and processing of RNA in Escherichia coli. The enzymatic activity of RNase E is controlled by the protein inhibitors RraA and RraB. The marine pathogenic bacterium Vibrio vulnificus also contains homologs of RNase E and RraA, designated as RNase EV, RraAV1, and RraAV2. Here, we report that RraAV1 actively inhibits the enzymatic activity of RNase EV in vivo and in vitro by interacting with the C-terminal domain of RNase EV. Coexpression of RraAV1 reduced ribonucleolytic activity in the cells overproducing RNase EV and consequently restored normal growth of these cells. An in vitro cleavage assay further demonstrated that RraAV1 efficiently inhibits the ribonucleolytic activity of RNase EV on BR10 + hpT, a synthetic oligonucleotide containing the RNase E cleavage site of RNA I. Our findings suggest that RraAV1 plays an active role in RNase EV-mediated RNA cleavage in V. vulnificus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Control of RNA stability plays a critical role in determining the transcript levels and contributes to the regulation of gene expression in the cell. In Escherichia coli, RNase E (Rne) is an essential endoribonuclease that controls the decay and processing of a large proportion of RNA in the cell [2, 3, 6, 8, 10, 11, 15, 19].
The 118-kDa RNase E, encoded by the rne gene, can be divided into distinct halves. The highly conserved N-terminal domain (amino acid residues 1–529) exhibits site-specific endoribonuclease activity that is essential for cell viability [9, 17]. The unstructured C-terminal domain (530–1061) provides a platform for the degradosome complex, which includes the polynucleotide phosphorylase (PNPase), RhlB RNA helicase, ATP-generating enzyme enolase, chaperone proteins DnaK and GroEL, and polyphosphate kinase [1].
The enzymatic activity and cellular concentration of RNase E are tightly controlled in E. coli through several mechanisms. Forced depletion or adventitious overexpression has been shown to cause cellular toxicity leading to growth retardation [4, 11]. RNase E maintains its relatively stable level by cleaving the 5′-untranslated region of its own transcript when RNase E activity exceeds cellular needs [7, 18, 20]. The endonucleolytic activity of RNase E is also regulated by the stoichiometric binding of the inhibitor proteins RraA or RraB to separate sites within the C-terminal domain of RNase E, which affects the composition of the degradosome [2, 5, 12]. A recent study also revealed that RraA-like proteins found in some bacterial species have divalent metal ion-dependent 4-hydroxy-4-methyl-2-oxoglutarate (HMG)/4-carboxy-4-hydroxy-2-oxoadipate (CHA) aldolase activity [16].
The marine pathogenic bacterium Vibrio vulnificus contains genes that encode orthologs of E. coli RNase E as well as two RraA-like proteins, designated as RNase EV (86.4 % amino acid sequence similarity with RNase E), RraAV1, and RraAV2 (80.1 and 59 % amino acid sequence similarity, respectively, with RraA). These homologs of RraA have been shown to inhibit E. coli RNase E differently [13, 14].
In this study, using a genetic system that takes advantage of the ability of RNase EV to functionally complement RNase E in rne-deleted E. coli cells, we investigated the ribonucleolytic effect of RraAV1 on RNase EV and its mechanism of action.
Materials and Methods
Strains and Plasmids
Plasmids pKAN6B and pKAN6B-RraAV1 have been described previously [13]. Plasmids pLAC-RNEV2 and pNRNEV5 were constructed by ligating polymerase chain reaction (PCR)-DNA digested with NotI and SphI restriction enzymes into the same restriction enzyme sites in pLAC-RNE2. We amplified DNA fragments containing regions that encode full-length and N-terminal sequences of RneV with hexahistidine tags at the C-termini using the same primers, RNEV-F (5′-CACAGCGGCCGCAGGAGGTTACGA-3′) and RNEV-R (5′-CAGTGCATGCCTAGTGGTGGTGGTGGTGGTGGTGGTG-3′) with pRNEV4 and pNRNEV4 as the respective templates.
Measurement of Plasmid Copy Number
The cells were grown to an OD600 of 0.1 in the presence of 10 μM isopropylthiogalactoside (IPTG) and 0.2 % arabinose. Then, 0 or 1 mM IPTG was additionally added to the cultures, which were further grown to an OD600 of 1.5 and harvested to obtain plasmid DNA. Plasmids were digested with the NotI restriction enzyme, which is a site that is unique in pLAC-RNEV2 and pNRNEV5, as well as in pKAN6B and pKAN6B-RraAV1. The digested plasmid DNA was electrophoresed in 0.8 % agarose gel and stained with ethidium bromide. The plasmid copy number was calculated relative to the concurrently present pSC101 derivatives (pLAC-RNEV2 and pNRNEV5), which undergo replication independent of RNEV, by measuring the molar ratio of the pSC101 derivatives to the ColE1-type plasmids (pKAN6B and pKAN6B-RraAV1).
Protein Expression and Purification
DK001 and DK002 cells were transformed with pKAN6B-RraAV1 and cultured to an OD600 of 0.1 in the presence of 10 μM IPTG and 0.2 % arabinose, and 1 mM IPTG was added to the cultures. The cells were further grown to an OD600 of 1.5 and harvested for copurification studies. Cells were broken by French Press followed by binding of the hexahistidine-tagged RneV and N-RneV on Ni–NTA agarose beads (Invitrogen) overnight. The beads were then washed four times with lysis buffer, and the proteins were eluted with 400 mM imidazole buffer. RraAV1 was detected using rabbit polyclonal antisera against RraA (1:500 dilution). Purified RraAV1 Protein was obtained from Dr. Nam-Chul Ha in Seoul National University, Republic of Korea.
In Vitro Cleavage of RNase E Substrate
BR10 + hpT was labeled with [γ-32P]-ATP at the 5′-end using T4 polynucleotide kinase (Takara, Japan), and the labeled products (p-BR10 + hpT) were purified using MicroSpin™ G25 columns (GE Healthcare, UK) according to the manufacturer’s instructions. Approximately, 0.5 pmol of p-BR10 + hpT was pre-incubated with 2 pmol of purified RneV or 4 pmol of N-RneV proteins with varying concentrations of RraAV1 on ice for 10 min in 20 μl of 200 mM Tris–HCl buffer (pH 8.0) containing 1 M NaCl, 1 mM DTT, 50 mM MgCl2, and 50 % (v/v) glycerol. The cleavage reaction proceeded at 37 °C for 2 h. The reaction products were separated by electrophoresis using 12 % denaturing polyacrylamide gel.
Results and Discussion
Effects of Coexpression of RraAV1 on the Growth of E. coli Cells Overproducing RneV Proteins
We wished to resolve the question of whether V. vulnificus RraAV1 can modulate the ribonucleolytic activity of RNase EV (RneV). Based on our previous finding that RNase EV can functionally complement RNase E in E. coli [14], we developed a genetic system that uses an rne-deleted E. coli strain expressing RNase EV. The coding regions of the full-length and N-terminal sequences of RneV (N-RneV) with hexahistidine tags at the C-termini were cloned into pLAC-RNE2 by replacing the rne gene encoding RNase E under the control of the IPTG-inducible lacUV5 promoter. The resulting plasmids, pLAC-RNEV2 and pNRNEV5, were used to transform E. coli strain KSL2000, which contains a deletion in the chromosomal rne gene that is complemented by a plasmid-borne rne gene under the control of an arabinose-inducible promoter (pBAD-RNE). Introduction of an incompatible ampicillin-resistant (Apr) plasmid expressing RneV or N-RneV (pLAC-RNEV2 and pNRNEV5) into the KSL2000 strain and selection of the incoming plasmid by growing transformants containing both plasmids (pBAD-RNE and pLAC-RNEV2 or pNRNEV5) in the presence of ampicillin (100 μg/ml) and 10 μM IPTG for 40 generations resulted in the displacement of the resident Kmr plasmid by the Apr RneV-expressing construct (DK001) or N-RneV-expressing construct (DK002).
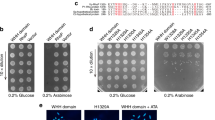
First, we investigated the growth characteristics of DK001 and DK002 cells. The growth of DK001 cells was inhibited by overexpression of RneV in the presence of 1 mM IPTG, whereas overexpression of N-RneV did not affect the growth of DK002 cells. This phenomenon has been observed in rne-deleted E. coli cells that express the full-length or N-terminal catalytic domain of Rne [10], suggesting that RneV proteins possess ribonucleolytic activities that are similar in extent and specificity to those of Rne proteins. Next, to test the effect of RraAV1 on RNase EV activity, a compatible Kmr plasmid expressing RraAV1 under the control of the arabinose-inducible promoter (pKAN6B-RraAV1) was introduced into DK001 and DK002 cells. DK001 cells overproducing both RneV and RraAV1 proteins in the presence of 1 mM IPTG and 0.2 % arabinose were able to grow at rates similar to those of the DK001 cells harboring an empty vector (pKAN6B) and grown in the medium containing 10 μM IPTG and 0.2 % arabinose (Fig. 1). Coexpression of RraAV1 and N-RneV in DK002 cells did not affect the growth rate of these cells (Fig. 1). These results imply that these V. vulnificus proteins have physiological activities in E. coli that are functionally equivalent to RNase E and RraA; RraAV1 effectively modulates the enzymatic activity of RneV for proper processing and decay of RNA species required to restore normal cellular growth in RneV-overproducing E. coli cells.

Effects of overproduction of RNase EV and coexpression of RraAV1 on E. coli growth. a Coexpression of RneV (Black) or N-RneV (Gray) and RraAV1 procedure. Promoters for plasmids (pBAD, placUV5), replication origin of plasmids (pSC101 ori, p15A ori), and antibiotic markers (Ap r, Km r) are indicated. b Effects of coexpression of RraAV1 in E. coli cells overproducing RNase EV on growth. The cultures of DK001 or DK002 cells harboring pKAN6B or pKAN6B-RraAV1 were grown in LB-10 μM IPTG medium, 0.2 % arabinose, and no additional IPTG (DK001 or DK002 + pKAN6B + 10 μM IPTG) or 1 mM IPTG (DK001 or DK002 + pKAN6B + 1 mM IPTG and DK001 or DK002 + pKAN6B-RraAV1 + 1 mM IPTG) was added to the cultures at OD600 = 0.1, and growth was monitored by analyzing cell density (absorbance at 600 nm) at time intervals. c Western blot analysis of cultures obtained from the growth curve. Culture samples from (b) were harvested in the log phase (OD600 = 1.5) to obtain total protein. The same membrane probed with anti-His monoclonal antibody and anti-RraA polyclonal antibody was tripped and reprobed with anti-S1 polyclonal antibody
Effects of Coexpression of RraAV1 on Ribonucleolytic Activity of RNase EV In Vivo
To test whether RraAV1 is able to modulate the ribonucleolytic activity of RneV in vivo, four of the known Rne substrates in E. coli were analyzed for their steady-state level. The first substrate tested was RNA I, which acts as an antisense repressor of ColEI-type plasmid replication. The in vivo activity of RneV against RNA I was assessed by measuring the relative copy number of a ColE1-origin plasmid (pKAN6B or pKAN6B-RraAV1) to a pSC101-origin plasmid (pLAC-RNEV2) in E. coli DK001 cells when RraAV1 is conditionally expressed by arabinose. Forced overexpression of RneV in the presence of 1 mM IPTG in DK001 cells increased the copy number of the ColE1-type plasmid pKAN6B by approximately 2.5-fold relative to that observed in cells expressing RneV in the presence of 10 μM IPTG. Increased ColE1-type plasmid copy number with forced overexpression of RneV in the presence of 1 mM IPTG was reduced to levels similar to that of the cells expressing RneV in the presence of 10 μM IPTG when RraAV1 was coexpressed in the presence of 0.2 % arabinose. These results indicate that RraAV1 effectively inhibited RNase EV action on RNA I in vivo (Fig. 2a, left panel). The inhibitory activity of RraAV1 on N-RneV appeared to be moderate, as indicated by a moderate decrease in the copy number of the ColE1-type plasmid pKAN6B-RraAV1 compared to that of pKAN6B in DK002 cells overexpressing N-RneV (Fig. 2a, right panel). The moderate inhibitory effect of RraAV1 on N-RneV might have resulted from the fact that its in vivo ribonucleolytic activity is significantly lower than that of the full-length RneV, as has been shown for E. coli N-Rne. Normal growth rates of DK002 cells overproducing N-RneV support this hypothesis. Another possibility is that RraAV1 requires the C-terminal domain to efficiently interact with RneV, which has been shown for RraA.

Effects of coexpressed RraAV1 on the ribonucleolytic activity of RNase EV in vivo. a Effects of overproduced RneV or N-RneV and coexpressed RraAV1 on the copy number of ColE1-type plasmids. Plasmid DNA was isolated from cultures used in the growth curve analysis (Fig. 1b). Plasmids digested with the NotI restriction enzyme, which has a unique cleavage site in all plasmids tested here, were electrophoresed in 0.8 % agarose gel and stained with ethidium bromide. Plasmid copy number was calculated relative to the concurrently present pSC101 derivatives (pLAC-RNEV2 or pNRNEV5), the replication of which is independent of Rne, by measuring the molar ratio of the pLAC-RNEV2 or pNRNEV5 plasmids to ColE1-type plasmids (pKAN6B or pKAN6B-RraAV1). Densitometric measurements of bands corresponding to each plasmid were converted to actual ratios after normalizing the values according to the size of ColE1-type plasmids and are shown at the bottom of the gel. b Effects of overproduced RneV or N-RneV and coexpressed RraAV1 on the steady-state level of ftsZ, rnhB, and rpoS mRNA. Total RNA was isolated from DK001 or DK002 cells grown to an OD600 of 1.5 in the same way as described in Fig. 1b, and reverse transcriptase-polymerase chain reaction (RT-PCR) was performed. The relative abundance of each mRNA is shown at the bottom of the gels. The experiments were performed at least three times, and the standard error of the mean (±numbers) was used to indicate the range of the assay results in the graph. c Interactions of RNase EV with RraAV1. The cultures of DK001 and DK002 cells harboring pKAN6B or pKAN6B-RraAV1 were grown in LB-10 μM IPTG medium and 0.2 % arabinose, and 1 mM IPTG was added to the cultures at OD600 = 0.1. Culture samples were harvested in the log phase (OD600 = 1.5) to obtain total protein. The same membrane probed with anti-His monoclonal antibody and anti-RraA polyclonal antibody was tripped and reprobed with anti-S1 polyclonal antibody. His-tagged RneV or N-RneV in cells expressing RraAV1 was purified using Ni–NTA agarose resin, and the copurified RraAV1 was identified by Western blot analysis using anti-RraA polyclonal antibody
We further investigated the ribonucleolytic activity of RNase EV on other RNase E substrates when RraAV1 was coexpressed. The abundance of mRNA transcripts of the three genes, rpsO, rnhB, and ftsZ, was measured using semi-quantitative RT-PCR [13]. The results show that coexpression of RraAV1 efficiently inhibited RneV action on these mRNA transcripts in DK001 cells, resulting in the increased abundance of each RNA species in vivo, whereas no significant changes associated with RraAV1 coexpression were observed in DK002 cells (Fig. 2b).
Physical Interactions Between RNase EV and RraAV1
To elucidate the basis for the observed differential inhibitory effects of RraAV1 on RneV and N-RneV, physical interactions between these proteins were examined by copurification experiments. RraAV1 was coexpressed in DK001 and DK002 cells and C-terminal hexahistidine-tagged RneV and N-RneV were affinity-purified using Ni–NTA beads. When purified RneV and N-RneV proteins were analyzed for RraAV1 by Western blot analysis, we found that RraAV1 was highly copurified with RneV, than with N-RneV (Fig. 2c). These results showed that the C-terminal domain is required for high-affinity binding of RraAV1 to RneV, which explains why RraAV1 more effectively inhibited the ribonucleolytic activity of RneV than N-RneV in vivo.
Inhibition of the Catalytic Activity of RNase EV by RraAV1 In Vitro
We wished to examine whether the inhibitory effect of RraAV1 on the ribonucleolytic activity of RneV in vivo results from direct action of RraAV1 on RneV, which requires no other factor(s). For this reason, RraAV1 and the full-length RneV and N-terminal part of RneV were affinity-purified for in vitro cleavage assays. For an RNA substrate, we utilized a 5′-32P-end-labeled BR10 + hpT (p-BR10 + hpT), a synthetic oligonucleotide that contains the RNA I cleavage site of RNase E and surrounding nucleotides located at its 5′ end to sequences corresponding to the distal hairpin loop and transcription termination regions at the 3′ end of RNA I (Fig. 3a). Addition of RraAV1 to the reaction resulted in a high degree of inhibition on the ribonucleolytic activity of RneV in a dose-dependent manner (Fig. 3b), while RraAV1 was not able to effectively inhibit ribonucleolytic activity of N-RneV (Fig. 3c). These results show that RraAV1 inhibits the ribonucleolytic activity of RNase EV via its direct action on the catalytic activity of the enzyme, which requires the C-terminal part of RNase EV for efficient inhibition.

Effects of RraAV1 on the ribonucleolytic activity of RNase EV in vitro. a Secondary structure of BR10 + hpT RNA substrate deduced using the M-fold program. The RNase EV cleavage site is indicated by an arrow. b RraAV1 inhibition of p-BR10 + hpT cleavage by RNase EV in vitro. About 2 pmol of 5′-end-labeled p-BR10 + hpT RNA was incubated with 2 pmol of RneV with varying concentrations of RraAV1, 400 pmol of RraAV1, or 400 pmol of BSA in 20 μl of 1X cleavage buffer at 37 °C for 2 h for RneV, RraAV1 only, or BSA only controls. Samples were mixed with an equal volume of loading buffer, and then denatured at 65 °C for 5 min and loaded onto a 12 % polyacrylamide gel containing 8 M urea. The percentage of uncleaved p-BR10 + hpT in the gel was quantitated using a phosphorimager and OptiQuant software. c RraAV1 inhibition of p-BR10 + hpT cleavage by N-RneV in vitro. 2 pmol of 5′-end-labeled p-BR10 + hpT RNA was incubated with 4 pmol of N-RNase EV with varying concentrations of RraAV1, 400 pmol of RraAV1, or 400 pmol of BSA in 20 μl of 1X cleavage buffer at 37 °C for 2 h for N-RneV, RraAV1 only, or BSA only controls. Inhibition rates were determined by measuring the percentage of the remaining p-BR10 + hpT, as described previously
In recent years, the importance of RNase E in the regulation of gene expression via RNA degradation and processing has been extensively studied and many details on mechanisms to modulate its ribonucleolytic activity have been unveiled in E. coli. Our findings indicate that RNase EV and RraAV1 have physiological activities in E. coli that are unequivocally functionally equivalent to RNase E and RraA, implying that RraAV1 plays an active role in RNase EV-mediated RNA cleavage in V. vulnificus. RraAV2, the other V. vulnificus homolog of RraA, did not show significant physiological activities on RNase EV in E. coli (data not shown), suggesting that its mode of action on RNase EV is likely to be different from that of RraAV1. Considering the degrees of RraAV1 and RraAV2 homology to RraA and their differential inhibitory effects on RNase E and RNase EV in E. coli [13], it is tempting to speculate that binding of RraAV1 and RraAV2 to RNase EV results in alteration of the enzyme’s substrate specificity in V. vulnificus. Further studies will reveal their physiological role in this pathogenic marine bacterium.
References
Callaghan AJ, Aurikko JP, Ilag LL et al (2004) Studies of the RNA degradosome-organizing domain of the Escherichia coli ribonuclease RNase E. J Mol Biol 340:965–979
Gao J, Lee K, Zhao M et al (2006) Differential modulation of E. coli mRNA abundance by inhibitory proteins that alter the composition of the degradosome. Mol Microbiol 61:394–406
Ghora BK, Apirion D (1978) Structural analysis and in vitro processing to p5 rRNA of a 9S RNA molecule isolated from an rne mutant of E. coli. Cell 15:1055–1066
Go H, Moore CJ, Lee M et al (2011) Upregulation of RNase E activity by mutation of a site that uncompetitively interferes with RNA binding. RNA Biol 8:1022–1034
Gorna MW, Pietras Z, Tsai YC et al (2010) The regulatory protein RraA modulates RNA-binding and helicase activities of the E. coli RNA degradosome. RNA 16:553–562
Hagege JM, Cohen SN (1997) A developmentally regulated Streptomyces endoribonuclease resembles ribonuclease E of Escherichia coli. Mol Microbiol 25:1077–1090
Jain C, Belasco JG (1995) Autoregulation of RNase E synthesis in Escherichia coli. Nucleic Acids Symp Ser 33:85–88
Jain C, Deana A, Belasco JG (2002) Consequences of RNase E scarcity in Escherichia coli. Mol Microbiol 43:1053–1064
Kido M, Yamanaka K, Mitani T et al (1996) RNase E polypeptides lacking a carboxyl-terminal half suppress a mukB mutation in Escherichia coli. J Bacteriol 178:3917–3925
Lee K, Bernstein JA, Cohen SN (2002) RNase G complementation of rne null mutation identifies functional interrelationships with RNase E in Escherichia coli. Mol Microbiol 43:1445–1456
Lee K, Cohen SN (2003) A Streptomyces coelicolor functional orthologue of Escherichia coli RNase E shows shuffling of catalytic and PNPase-binding domains. Mol Microbiol 48:349–360
Lee K, Zhan X, Gao J et al (2003) RraA. A protein inhibitor of RNase E activity that globally modulates RNA abundance in E. coli. Cell 114:623–634
Lee M, Yeom JH, Sim SH et al (2009) Effects of Escherichia coli RraA orthologs of Vibrio vulnificus on the ribonucleolytic activity of RNase E in vivo. Curr Microbiol 58:349–353
Lee M, Yeom JH, Jeon CO et al (2011) Studies on a Vibrio vulnificus functional ortholog of Escherichia coli RNase E imply a conserved function of RNase E-like enzymes in bacteria. Curr Microbiol 62:861–865
Li Z, Deutscher MP (2002) RNase E plays an essential role in the maturation of Escherichia coli tRNA precursors. RNA 8:97–109
Mazurkewich S, Wang W, Seah SY (2014) Biochemical and structural analysis of RraA proteins to decipher their relationships with 4-hydroxy-4-methyl-2-oxoglutarate/4-carboxy-4-hydroxy-2-oxoadipate aldolases. Biochemistry 53:542–553
McDowall KJ, Cohen SN (1996) The N-terminal domain of the rne gene product has RNase E activity and is non-overlapping with the arginine-rich RNA-binding site. J Mol Biol 255:349–355
Mudd EA, Higgins CF (1993) Escherichia coli endoribonuclease RNase E: autoregulation of expression and site-specific cleavage of mRNA. Mol Microbiol 9:557–568
Ow MC, Liu Q, Kushner SR (2000) Analysis of mRNA decay and rRNA processing in Escherichia coli in the absence of RNase E-based degradosome assembly. Mol Microbiol 38:854–866
Sousa S, Marchand I, Dreyfus M (2001) Autoregulation allows Escherichia coli RNase E to adjust continuously its synthesis to that of its substrates. Mol Microbiol 42:867–878
Acknowledgments
This research was supported by the National Research Foundation of Korea (2014R1A2A2A09052791) and Chung-Ang University Research Scholarship Grants in 2013. We thank Drs. Saemee Song and Nam-Chul Ha for providing us with purified RraA1 protein.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Daeyoung Kim and Yong-Hak Kim have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Kim, D., Kim, YH., Jang, J. et al. Functional Analysis of Vibrio vulnificus Orthologs of Escherichia coli RraA and RNase E. Curr Microbiol 72, 716–722 (2016). https://doi.org/10.1007/s00284-016-1007-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-016-1007-y