
Abstract
Understanding diversity and distribution patterns of fungi, including yeasts, ultimately depends on accuracy of species recognition. However, different approaches to yeast species recognition often result in different entities or operational taxonomic units. We studied the effects of using different yeast species recognition approaches, namely morphological species recognition (MSR) and phylogenetic species recognition (PSR), on the distribution patterns of widespread basidiomycetous yeasts. Hence, we have revised a collection of yeast fungi isolated from spatially remote birch forests in the Moscow Region and Western Siberia with molecular typing and identification tools. PCR fingerprinting and rDNA sequencing analyses of strains of nine species previously identified on the basis of morphological and physiological tests (MSR) yielded 21 phylogenetic species (PSR), including three currently undescribed taxa. The number of distinct phylogenetic species comprised within a single morphospecies ranged from one to seven. A total of ten species were found in both regions, whereas the distribution of 11 yeasts was restricted to a single region only. Both geographical region and type of substrate (plant or soil) influence yeast distribution. Cryptococcus wieringae, C. victoriae, C. magnus, and Leucosporidium scottii were frequently found on plant substrates, whereas C. terricola and C. podzolicus were associated to soil substrates. Occurrence of C. magnus, C. albidus and Sporobolomyces roseus was found to depend on the geographical region. Microsatellite-PCR fingerprinting, MSP-PCR, applied to studying yeast intraspecific variability revealed three different types of distribution: (a) variability that depends on geographical factors (Curvibasidium cygneicollum, C. podzolicus, C. victoriae), (b) genetic identity irrespectively of the region of isolation (Rhodotorula pinicola, C. terricola), and (c) high degree of genetic variability that did not correlate with region of sampling (C. albidus and C. magnus).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Yeasts are reported to be inhabitants on different kinds of plant substrates, whether living or decaying, in different biotopes of many regions [2, 12]. A particular approach to yeast ecology has been undertaken by Bab’eva et al.: instead of focusing on specific substrates, i.e. plant species, they have chosen to analyze the spatial structure and biogeography of yeast populations obtained from large numbers of samples in the plant–soil system (phylloplane, litter and soil, e.g. [2, 34]). The structure of yeast communities changes throughout different geographical zones, from tundra to desert, when determined according to their phenotypic characteristics [6]. Along the latitudinal gradient, both higher yeast densities and species richness were found in boreal forest biotopes, for which some differences in community composition were noted depending on the forest type, spruce, alder or birch [34]. However, it was difficult to conclude whether the observed differences are solely determined by the vegetation type of or just by spatial separation. Therefore, a survey of yeast communities associated with a single forest type, birch forests, in two spatially, geographically, separated regions was performed [60]. The analysed forests in Moscow region (studied by Maksimova and Chernov [34]) and in Novosibirsk (Western Siberia) presented similar climatic conditions (average summer and winter temperatures, rainfall), soil type, and vegetation (dominating plant species). Yeast communities as determined from phenotypic keys [4, 27] were notably alike, even though the two sampling regions were 3,500 km apart. Yeast abundance and composition were found to depend on the type of substrate, namely live plant parts, litter and soil. Phylloplane yeasts were mostly represented by Cryptococcus albidus, C. laurentii, C. terricola, C. podzolicus, Rhodotorula glutinis, R. minuta and Sporobolomyces roseus which were found in the same abundance in both regions. Topsoil was inhabited by C. terricola and C. podzolicus. A single species, C. diffluens, was significantly more abundant in Western Siberia, particularly in soils.
Molecular identification methods have mainly replaced physiological assimilation tests to recognise distinct species on yeast diversity studies, as commonly used assimilation tests are not necessarily able to distinguish between closely related species. In fact, the use of molecular markers for taxa differentiation has vastly increased the number of novel recognised yeast species in the last decade, e.g. [28, 29, 44]. Yet, the mechanisms determining distribution of microorganisms, and yeasts in particular, in their natural habitats are still poorly understood. This is partly due to the complexity of some systems, like soil and phyllosphere, which limits the possibility to uncover mechanisms influencing abundance, composition, structure and distribution patterns of microbial communities, e.g. [2, 5, 24, 62].
Due to the small cell size, microorganisms were long proposed to have boundless dispersal abilities and, consequently, the development of microbial populations was assumed to be rather limited by the environment than spatial barriers, e.g. [24]. However, distinct biogeographical patterns have been demonstrated for bacteria [55] and fungi [33, 48]. Also, Taylor et al. [48] have demonstrated that the distribution range of a fungal species may depend upon the method of species recognition, morphological (MSR), biological (BSR) and phylogenetic (PSR). While some morphologically defined fungal species showed global distribution, species defined by phylogenetic species recognition approaches typically showed smaller geographical ranges with many endemic species. Despite the growing number of studies using PSR approaches to identify yeast species from the environment, still little is known about the distribution patterns and ranges of many phylogenetic species. In contrast, widespread occurrence of yeast species identified with MSR has been extensively reported.
In this study, we assessed the effects of spatial factors on yeast communities inhabiting phylloplane and soil substrates. We report on the PSR reassessment of MSR data on the distribution of dominating basidiomycetous yeast species in two geographically separated forests in Moscow and Novosibirsk (Western Siberia) regions, using microsatellite-PCR fingerprinting and rDNA sequencing strategies. We compared the results obtained using both MSR and PSR approaches for yeast identification in order to: (i) evaluate the effect of geographical barriers on yeast species distribution and population structure; (ii) compare the ecology of identified species with previous surveys that also used molecular identification methods; and (iii) propose updated hypotheses regarding previous studies that only employed phenotypic characterisation of the yeast isolates.
Materials and Methods
Sample Collection
Samplings were carried out in the month of August of three consecutive years (2002–2004). Studied biotopes were located in two geographically isolated regions of Russia, namely Western Siberia and Moscow region. Climatic conditions of these regions are similar as they are located at nearly the same latitude. Main sampling sites were situated in Novosibirsk (approximate coordinates 54.86N, 83.05E) and western part of Moscow region, near Burtsevo settlement (approximate coordinates 55.98N, 33.60E). During August 2003, samples were collected in Western Siberia, in Novosibirsk and at the border of the Altay State Natural Reserve (approximate coordinates 51.34N, 87.78E). During August 2004, sampling was carried out mainly in Moscow region, i.e. Burtsevo, Biological station “Malinki” of A.N. Severtsov Institute of Ecology and Evolution RAS (approximate coordinates 55.46N, 37.18E) and at Losiny Ostrov National Park (approximate coordinates 55.85N, 37.75E).
Samples were collected in mixed birch forests with main stands of Betula verrucosa (together with B. alba in Siberia) and Sorbus aucuparia (S. sibirica in Siberia). The herbs Fragaria vesca, Deschampsia cespitosa, Potentilla spp., Stellaria media, Aegopodium podagraria, and Equisetum sylvaticum were common to the two studied regions. Coniferous trees found regularly in birch forests, namely Pinus sylvestris (P. sibirica, in Siberia) and Picea abies (in European Russia) were also sampled. All plants were sampled in equal proportions during each sampling survey. In addition, a few other plants were exclusively collected in Moscow region, Acer spp., Aesculus hippocastanum, Swida spp., Corylus avellana, Dactylis glomerata, and Taraxacum officinale as they were observed growing in mixed birch forests. Plant substrates, i.e. green leaves, dried plant material and litter, and soil were collected in the corners of five randomly selected 100 × 100 m plots. Each of the four composite samples (50–200 g) collected per plot was composed of material derived from a single plant species (but different individuals), with green leaves, dry leaves and litter sampled and analysed separately. All composite samples were prepared in the field. For soil samples, coarse woody debris, roots and stones larger than 5 mm were removed in the field.
Yeast Isolation
From each composite sample, three to five sub-samples were randomly taken for yeast isolation. Yeast cultures were isolated by the conventional plating method, in which leaf washings and soil suspensions were plated onto solid medium. Substrate samples were placed in 100 ml glass flasks, suspended 1:10 to 1:50 (w/v) in sterile demineralised water and crushed with tissue homogeniser (3,000 rpm, 3 min). Aliquots of 0.1–0.2 ml of each sample suspension were plated on the surface of malt agar medium acidified with 40 % lactate (final pH 4.0–4.5). Each sample was plated in triplicate. Plates were incubated at room temperature for 2–3 days and then kept at lower temperatures, 6–10 °C, to prevent fast development of moulds. Plates were examined after 7, 14 and 21 days of incubation. Colonies were differentiated into macro-morphological types using dissection microscopy, counted, and 1–2 representatives of each colony type per plate were transferred into pure culture. Strains were maintained on the same medium as slants at 4 °C. Strains used in this study are listed in Table 1. Reference yeast strains were obtained from the Centraalbureau voor Schimmelcultures (CBS, Utrecht, The Netherlands) and the Portuguese Yeast Culture Collection (PYCC, Caparica, Portugal). A few additional cultures representing widespread phenotypic species from older studies [2, 7, 34] were obtained from KBP culture collection (Moscow State University, Russia).
Strains Selection
This study was aimed to reassess the identification of yeasts previously identified using the morphological species recognition approach (MSR) as Cryptococcus laurentii, C. albidus, C. diffluens, C. terricola, C. podzolicus, Rhodotorula minuta, R. fujisanensis, R. glutinis, and Sporobolomyces roseus isolated in two remote regions of Russia and originally studied by Yurkov et al. [60]. Because not all original cultures analysed by Yurkov et al. [60] were available for this study, further sampling efforts took place at the years 2002–2003, to re-sample yeasts in Western Siberia, and at 2004, mainly focused on isolating cultures in the Moscow region. The isolates used in this study have been chosen to represent cultures isolated in both geographical regions from the main plant species (Table S1). Our selection of cultures was partly based on the physiological profiles formerly determined by Yurkov et al. [60]. Using this preliminary grouping, we reduced the number of strains selected for molecular characterization studies by taking one to three isolates of each phenotypic species from the same substrate or plant species.
Assimilation profiles were determined according to Yarrow [57] and utilisation of six aldaric acids and 11 aromatic compounds was tested for members of the genus Cryptococcus according to Fonseca [11] and Sampaio [38]. Strains identified as Rhodotorula fujisanensis were tested for compatibility with the mating type strains of Curvibasidium cygneicollum. Mating experiments were performed as described by Sampaio et al. [43].
Microsatellite-PCR Fingerprinting
Genomic DNA extraction and microsatellite-PCR fingerprinting (MSP-PCR) using primers M13 (5′-GAG GGT GGC GGT TCT-3′), (GAC)5 and (GTG)5 followed the protocols described by Sampaio et al. [42]. Gel electrophoresis images were acquired with the BioRad Gel Image system (Richmond, USA) and analyzed with GELCOMPAR, version 4.1 (Applied Maths, Belgium). Dendrograms were computed using the UPGMA clustering method. Ordination of the amplified DNA fragment profiles was performed using principal components analysis implemented in the Statistica 6.0 software (StatSoft Inc., USA) and is based on similarity matrix produced using bands position (length) information. In agreement with other studies, isolates demonstrating very high similarity (>90 %) of microsatellite electrophoretic profiles and identical physiological properties are believed to be conspecific, e.g. [22, 23]. Therefore, only one representative strain per microsatellite profile was used for further identification by rDNA sequencing.
DNA Sequence Analysis
PCR amplification prior to sequencing employed the forward primer ITS5 (5′-GGA AGT AAA AGT CGT AAC AAG G) and the reverse primer LR6 (5′-CGC CAG TTC TGC TTA CC) using a Uno II Thermal Cycler (Biometra, Germany) and the resulting amplicons were purified with the GFX Band Purification Kit (Amersham Biosciences). Cycle sequencing of the D1/D2 variable domains of the 26S rDNA employed the forward primer NL1 (5′-GCA TAT CAA TAA GCG GAG GAA AAG) and the reverse primer NL4 (5′-GGT CCG TGT TTC AAG ACG G) and that of the ITS region employed forward primer ITS1 (5′-TCC GTA GGT GAA CCT GCG G) and reverse primer ITS4 (5′-TCC TCC GCT TAT TGA TAT GC), following protocols described by Inácio et al. [22, 23]. Sequences were obtained with an ALFexpress II DNA Analyser (Amersham Pharmacia Biotech), according to the manufacturer’s instructions.
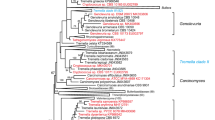
Alignment of nucleotide sequences was performed by the MAAFT algorithm [26]. Phylogenetic trees were computed using the maximum likelihood method with RAxML, version 7.0.3, and isolates’ placement was further validated with 1,000 rounds of bootstrap replicates [46]. Model of DNA substitution (GTR) and parameters for maximum likelihood analysis were derived from Modeltest, version 3.7 [37]. Nucleotide sequences were deposited in GenBank under the accession numbers given in the phylogenetic trees (Figs. 1, 2 and 3). Additional sequences were retrieved from GenBank and CBS databases (accession numbers and strain numbers are indicated on the phylogenetic trees).

Phylogenetic tree of selected isolates obtained by maximum likelihood analysis of 26S rRNA gene (D1/D2 domains). The numbers given on branches are frequencies (>50 %) with which a given branch appeared in 1,000 bootstrap replications. Sequences determined in the present study are typed in boldface. The scale indicates the number of expected substitutions accumulated per site. Names of the clades are given according to Fonseca et al. (2011)

Phylogenetic placement of isolates related to C. victoriae obtained by maximum likelihood analysis of 26S rRNA gene (D1/D2 domains). Nucleotide sequences with missing GenBank accession numbers were retrieved from CBS database. Other details as for Fig. 1

Phylogenetic placement of Cryptococcus species in the Filobasidiales lineage obtained by maximum likelihood analysis of ITS region sequences. Nucleotide sequences with missing GenBank accession numbers were retrieved from CBS database. Names of the clades are given according to Fonseca et al. (2011). Other details as for Fig. 1
Statistical Analysis
For each sub-sample, yeast quantity and community structure were determined. Yeast quantity was calculated as colony-forming units (CFU) per gram of dry plant material or soil at natural humidity. The community structure was characterised by relative abundance and frequency of occurrence of every observed species in the sample, see [62]. Distribution of basidiomycetous yeasts was analysed using previously published data referring to sampling surveys 1998, 2001, and 2002 [60], and results obtained in this study, sampling years 2003–2004. We analysed yeast communities composition (based on species’ presence or absence) and structure (based on relative abundance values) extrapolating identification results (PCR fingerprinting and rDNA sequencing) on sub-samples, for which it was possible to attribute molecular identification results to the original yeast cultures or groups of cultures (Table S1) re-identified in the current study. Because not all phenotypically characterised cultures were available for the reassessment, we substantially reduced the original dataset, which was based on phenotypic identification results (MSR) and contained information of 200 sub-samples (sampling years 2003–2004) and 150 sub-samples (sampling 2002) analysed previously by Yurkov et al. [60]. After the reassessment of the data on the distribution of analysed basidiomycetous yeast species, the dataset was reduced to 248 sub-samples, most of which were sampled in 2002-2004 (Table S1).
The influence of spatial and environmental factors on the yeast communities’ structure was analysed by ANOVA/MANOVA block implemented in the Statistica 6.0–9.0 software (StatSoft Inc., USA). Statistical analyses were performed to assess effects of geographical region (Western Siberia and Moscow region), and substrate (plant material and soil). Effects of a particular plant species were analysed for the plants that were common to both regions, namely Betula spp., B. verrucosa (and B. alba), Sorbus spp., S. aucuparia (and S. sibirica), Fragaria vesca, Deschampsia cespitosa, Aegopodium podagraria and Equisetum sylvaticum. The homogeneity of variance was assessed with Levene’s test. Distribution of all dependent variables was close to normal, unimodal but asymmetric in some cases. Results of multivariate analysis of variance, MANOVA, were confirmed by ANOVA analyses. Effects were considered to be statistically significant at the level P ≤ 0.05. Significant effects were additionally confirmed with χ 2 test.
Results
Species Inventory
A total of 100 isolates were studied and out of them a total of 57 strains were selected for sequence-based identification. The following 17 yeast species were identified using PCR fingerprinting (results not shown) and rDNA sequencing (Table 1; Figs. 1, 2 and 3): Cryptococcus victoriae, C. carnescens, C. foliicola, C. heimaeyensis, C. tephrensis (victoriae clade), and C. podzolicus in the Tremellales; C. magnus, C. wieringae, C. oeirensis (floriforme clade), C. albidus, C. albidosimilis, C. adeliensis (albidus clade), and C. terricola in the Filobasidiales; Sporobolomyces roseus and Rhodosporidium babjevae, in the Sporidiobolales; Rhodotorula pinicola and R. slooffiae, in the Cystobasidiales and Curvibasidium cygneicollum and Leucosporidium scottii in the Microbotryomycetes. Two isolates, AY-59 and AY-83 showed only low similarity to known species and may thus represent novel taxa (Fig. 1). Specifically, AY-59 was a close relative of Cryptococcus spp. CBS 7890 and CBS 8630 (Fig. 1), showing 9 and 2 nucleotide substitutions in the D1/D2 domains of the 26S rDNA gene, respectively. Isolate AY-83 was found to be related to C. aureus, but differed in more than 20 nucleotide positions in the D1/D2 domain from the latter species (Fig. 1). Among the strains available in public collections, AY-83 has the closest match with the strain Cryptococcus cf. aureus NRRL Y-30215 [10], showing only 3 indels in the D1/D2 domains. The number of phylogenetic species comprised within a single morphospecies ranged from 1 to 7 (Table 1). The use of a PSR approach revealed that 3, 4 and 7 distinct species were comprised within the most diverse MSR-based species C. diffluens, C. albidus and C. laurentii, respectively (Table 1).
Among dominating red-coloured yeasts, isolates that produced ballistoconidia were identified as S. roseus and those that fitted the standard phenotypic diagnosis of R. glutinis were found to represent Rhodosporidium babjevae (Table 1; Fig. 1). R. babjevae belongs to the Rhodotorula glutinis sensu stricto group, together with R. graminis [15]. Strains previously identified as R. minuta were found to represent two distinct closely related species, namely R. pinicola and R. slooffiae (Table 1; Fig. 1). Sequencing results confirmed the phenotypic identification of R. fujisanensis and showed that non-pigmented strains of Rhodotorula sp. belong to the species Leucosporidium scottii (Table 1; Fig. 1). Isolates representing R. fujisanensis were additionally studied using mating tests with reference strains of Cu. cygneicollum, the sexual state of R. fujisanensis [43]. The three strains of R. fujisanensis (AY-25, 27, and 28) mated with MAT A2 test culture and thus represent the opposite mating type of Cu. cygneicollum.
Phylogenetic placement of our isolates and strains from CBS collection revealed a complex structure of the victoriae clade (Fig. 2). This group comprises several currently recognised species [14], but also strains originally identified and presently listed in GenBank as Cryptococcus aff. victoriae. A single strain, AY-78, was identified in our study as C. foliicola [54] (Table 1; Fig. 2). Other isolate, AY-87, was identified as C. carnescens (Table 1; Fig. 2), with one nucleotide substitution from the type strain CBS 973. Phylogenetic analysis revealed two clusters of C. victoriae strains (Fig. 2). Isolates AY-92 and AY-99 showed D1/D2 sequences identical to those of the type strain of C. victoriae CBS 8685 (Fig. 2). A second group of strains (AY-68, 81, 82, 84, and 93) differs from the type strain of C. victoriae in two nucleotide positions, as well as CBS 8908 [49], CBS 9013 [20], and CBS 9206 [51] (Fig. 2). Isolate AY-97 presented four nucleotide substitutions in the D1/D2 region when compared to members of the two above-mentioned clusters and thus showed an isolated position in the phylogenetic tree (Fig. 2).
Similarly to C. victoriae, cultures of C. tephrensis analysed in this study formed two major groups. The first sub-cluster includes the isolates AY-94, 95, 100, and 103, which are closely related to the type strain of C. tephrensis CBS 8935. Isolate AY-77 grouped in the second sub-cluster with strains CBS 8993, CBS 9012, and CBS 9023 [20]; CBS 8934 and CBS 8968 [51]; and CBS 9799, isolated from a plant in Norway (according to CBS database), which have three nucleotide substitutions in the D2 domain from the type strain of C. tephrensis. Strains CBS 8935 and CBS 8934 are listed in the CBS database as type strains of distinct varieties, namely C. tephrensis var. tephrensis and C. tephrensis var. soli, respectively. However, these varieties have apparently not been validly described, see [14].
Physiological tests provided a good differentiation of the closely related species C. wieringae and C. magnus in the Filobasidiales lineage due to differences in assimilation of ribitol and d-glucosamine (data not shown). We were also able to distinguish C. tephrensis from other members of the C. victoriae clade. Our isolates of C. tephrensis did not assimilate mucic and saccharic acids as sole carbon sources, whereas other members of the C. victoriae clade demonstrated growth using these compounds. Data available at the CBS yeast database and the last revision of the genus Cryptococcus [14] confirm this observation since C. tephrensis assimilates these compounds weakly or not at all. However, physiological profiles of C. victoriae, C. carnescens, C. heimaeyensis and C. foliicola are very similar and, thus, do not provide good characters for differentiation.
Distribution and Ecology
During the yeast isolation, the number of yeast colonies assigned to a species identified using phenotypic characters (MSR) was recorded and expressed as a proportion of the total number of yeast colonies (or CFU) observed in a sub-sample. After the reassessment of species identification, we were able to track back strains origin and match relative abundance values with a phylogenetic (PSR) yeast species. Despite the limited number of strains re-identified in this study, each culture included in the study represented a sub-set of isolates identical on the basis of MSR approach, so we were able to included data from a total of 248 samples in the final dataset (Table S1). However, considering the limitation of our analyses, we present and discuss here about the average values of relative abundance and incidence reflecting the large-scale distribution patterns of basidiomycetous yeast species re-identified in the present study.
The relative yeast abundance data, average values at both sample collection regions and for each studied substrate, is reported in Table 1. The most frequently isolated yeast species in both regions was C. wieringae. Other frequently isolated species were C. victoriae, C. magnus and L. scottii. Cryptococcus terricola was the most frequent and abundant species found in soil samples. Statistical analyses showed that both type of substrate (plant vs. soil) and geographic region (Moscow vs. Western Siberia) are essential factors determining distribution of yeast species (Table 2). Asexual states of L. scottii and several pigmented basidiomycetes, S. roseus, R. babjevae, R. pinicola and R. slooffiae, were found on plant material only (Table 1). Members of the floriforme clade of Filobasidiales [14] were mostly restricted to live plant materials (Table 1; Fig. 3). While C. wieringae and C. magnus occurred frequently in the phylloplane, C. oeirensis was represented only by a single isolate in this substrate (Table 1; Fig. 3). Members of the victoriae clade of Tremellales [14] were frequent and abundant in plants and in litter (Table 1; Fig. 2). The majority of these isolates were identified as C. victoriae and C. tephrensis (Table 1). Three other species, C. carnescens, C. heimaeyensis, and C. foliicola were represented by single isolates only. Interestingly, for both of the above-mentioned clades, more than 90 % of the respective isolates belonged to two most frequent species pairs: a dominant (C. wieringae or C. victoriae) and a sub-dominant (C. magnus or C. tephrensis) species. Three strains isolated from live plants were identified as the closely related species C. adeliensis and C. albidosimilis (Table 1; Fig. 3). Soil samples were populated by C. terricola, C. podzolicus and C. albidus (Fig. 1). The last species was also observed on living plant parts collected in Moscow region (Table 1). Cu. cygneicollum was isolated from plant, litter and soil samples.
Geographical Signal in Populations & Intraspecific Variability
Next to the application of PSR approach for species identification, special attention was given to analyses of microsatellite-PCR fingerprinting profiles of the studied basidiomycetous yeasts in relation to geographical origin of the strains. Three different types of distribution were found: (a) variability that depends on geographical factors (Cu. cygneicollum, C. podzolicus, C. victoriae), (b) genetic identity independently of the region of isolation (C. terricola, R. pinicola), and (c) high degree of genetic variability that did not correlate with region of sampling (Filobasidiales: C. albidus and C. magnus).
The members of the first group formed clusters corresponding to the geographic origin (Fig. 4b). Strains of C. podzolicus displayed groupings both with (GTG)5 and M13 primers. Two reference strains of C. podzolicus were included in the study, as they were isolated from Moscow region, AY-36 (KBP 3365) and the type strain CBS 6819 (= PYCC 4488; KBP 6). It is interesting to note that the latter strain isolated by Bab’eva and Reshetova [3] and stored in collection for over 25 years grouped together with the other European isolates of C. podzolicus. Thus, the banding patterns revealed by MSP-PCR, which reflect genetic variability, appear to be stable over a long period of time. Similarly, strains of Cu. cygneicollum grouped according to their geographic origin (data not shown). Isolates identified as members of the victoriae clade (C. victoriae and C. tephrensis) showed diverse MSP-PCR fingerprint patterns. Quantitative comparison of the MSP-PCR profiles performed using ordination with principal component analysis (PCA) resolved a trend for separation of the isolates according to geographical origin and phylogenetic relatedness (Fig. 4c).

Examples of different intraspecific variability patterns observed in present study: a no variability (C. terricola), b geographical populations (C. podzolicus), c high variability with geographical trend (C. victoriae and C. tephrensis). a and b DNA banding patterns obtained with M13 primer and resulting dendrogram using Pearson’s coefficient and the UPGMA clustering method, co-phenetic correlation coefficient (r = 0.74). c DNA banding patterns obtained with (GAC)5 primer and resulting ordination using Pearson’s coefficient and principal components analysis (PCA). First two extracted components, factors describing together 36 % of the genetic variability between strains. A total of 75 % of data are described by six extracted components. Black markers designate isolates from Western Siberia and white ones from Moscow region. Strains of C. victoriae are marked with circles and those of C. tephrensis with squares. Large markers are centroids for groups according to geographical region of isolation
The second trend is represented by isolates showing low intraspecific variability as inferred by the highly similar microsatellite-PCR profiles with all primers. As an example, the similarity level among the C. terricola isolates was above 90 %, irrespectively of the isolation site (Fig. 4a). Similarly to isolates of C. terricola, all four strains of R. pinicola demonstrated almost identical profiles with (GAC)5 primer.
The third trend corresponds to strains belonging to the floriforme and albidus clades of Filobasidiales [14]. MSP-PCR revealed high levels of intraspecific variability, but the origin of the observed genetic divergence is unknown. Strains of C. albidus and its varieties formed two different groups that did not reflect the geographical origin, type of substrate or phylogenetic relationship. Similarly, isolates of the main dominating species C. wieringae demonstrated high levels of genetic variability that did not correlate with any of the analysed factors.
An ordination with PCA was found to be more appropriate for analysis of microsatellite-PCR profiles of the widespread phylloplane species, members of floriforme and victoriae clades, than conventional one-dimensional dendrograms. Due to high dissimilarity of profiles, no significant groupings were observed by direct analysis of the dendrograms. Ordination with PCA enables differentiation of fingerprints with at least two extracted components (axes). For example, the first two extracted components, factors describe together 36 % of the genetic variability between strains of C. victoriae and C. tephrensis, whereas the first component describes only 21 % alone (Fig. 4c). Thereby, ordination retains larger proportion of the information available for analyses than a one-dimensional dendrogram.
Discussion
Spatial Trends
On the community level, geographical region and type of substrate (phylloplane vs. soil) have been found to determine yeast species distribution, i.e. species richness and abundance (Table 2). Even though both factors significantly influence communities’ structure, effects of the substrate were found to be more pronounced in accordance with previous observations of Yurkov et al. [60]. Several species were only isolated from a single type of substrate (Table 1), which explains the difference between yeast communities in soils and live plant parts. Specifically, C. victoriae, C. magnus, C. wieringae, S. roseus, R. babjevae, Rh. pinicola, Rh. slooffiae and L. scottii are associated with live plant parts, whereas C. terricola, C. podzolicus and C. albidus are restricted to soil samples. In agreement with the results of Yurkov et al. [60] and regardless of the species recognition approach used, no association between yeasts species and individual plant species was found.
Upon utilizing the PSR approach, three significant differences in the phylloplane community reflecting spatial separation of the analysed forests were observed. Firstly, among the members of the floriforme clade, C. wieringae was isolated in both regions with similar abundance, whereas C. magnus was significantly more abundant in Western Siberia (Table 1). Secondly, the average relative abundance of C. albidus and its varieties was significantly higher in Western Siberia than European Russia, 5.1 and 0.4 %, respectively (Table 1). This trend in species distribution was also the only significant difference reported previously by Yurkov et al. [60]. Thirdly, the so-called “red yeasts” group appeared to consist of different species depending on the region studied, S. roseus in Moscow region and R. babjevae (not R. glutinis) in Western Siberia, respectively. Therefore, S. roseus might not have a wide occurrence in temperate zone (compare with [2, 6]), as it was not found in Western Siberia during this study. Also, opposing to the prevailing views on the worldwide distribution of R. glutinis, it was found that none of the studied strains belong to that species (Table 1, Table S1). Only five isolates are currently assigned to R. glutinis [39]. Similarly, R. graminis is known at this moment from eight isolates only [8, 39, 63].
On the population level, geographical signal varied considerably between different phylogenetic groups of species (Fig. 4). In general members of the Filobasidiales (C. albidus, C. terricola, C. wieringae and C. magnus) demonstrated high levels of genetic variability as determined by MSP-PCR that did not correlate with geographical region, type of substrate or phylogenetic position. In contrast, members of the Tremellales (C. podzolicus, C. victoriae) appeared to form geographical populations. To date, influence of spatial factors on yeast community composition in the same substrate type was demonstrated for simple and rather homogeneous substrates, like floral nectar [30], necrotic tissues of cacti [47], and berries [35]. Intraspecific variability that resulted from spatial separation of conspecific isolates has been previously observed in ascomycetous yeasts, for example Saccharomyces spp. [32, 33, 36], Candida sonorensis [16], and some other species [29, 31]. Previous analysis of ascomycetous yeasts Hanseniaspora guilliermondii, Torulaspora delbrueckii, and Debaryomyces hansenii isolated in Moscow region and Novosibirsk revealed distinct MSP-PCR fingerprint patterns with, (GTG)5 and (ATG)5 primers depending on the region of isolation, which demonstrated the existence of geographic populations [58]. Results obtained in the present study demonstrate that a similar pattern is apparently present not only within ascomycetous yeasts, but could be also observed among widespread basidiomycetes, namely C. podzolicus, C. victoriae and Cu. cygneicollum (Fig. 4).
Ecology and Distribution
Species concept has a great influence on diversity studies. Different approaches applied to species recognition may result in detecting different entities. A previous review aimed at resolving the impact of the species concept on biodiversity studies showed remarkable differences, with surveys based on a phylogenetic species concept detecting 48 % more species (300 % more for fungi) and an associated decrease in population size and range [1]. Although identification methods based on ribosomal DNA sequencing gained more and more importance in the last decade [48, 61, 62], some yeast ecologists repeatedly argued for using “natural” or “phenotypic” species in their studies as opposed to “genotypic” as the adaptation to environmental conditions results mainly from the phenotype, e.g. [17, 34]. However, these entities defined on morphological attributes (sometimes referred as sensu lato species) might not be restricted to a single phylogenetic lineage but include a broad set of unrelated taxa, e.g. [13]. Using a large collection of strains and different approaches to species identification, we discuss the phylogenetic relationships of yeasts comprised within the borders of species previously identified using only MSR approaches, namely C. laurentii, C. diffluens and C. albidus. Even though the selection strategy employed may not account for the full extent of species richness in these biotopes, it offers the possibility of re-assessment of species identification reported in earlier studies, e.g. [2, 17, 19, 34]. Thereby, we revise the distribution of identified phylogenetic species based on the comparison with earlier studies.
C. albidus, defined as a composite species complex, was considered an eurybiont species, as no significant trends in its distribution in time and space had been observed [2, 17, 19]. Our results suggest that mainly members of the floriforme clade (Filobasidiales, Tremellomycetes) are comprised within C. albidus (as identified with conventional physiological tests). Out of the two most frequent species in this group, C. magnus had been frequently reported from the phylloplane [12], but data regarding the distribution of the closely related species C. wieringae is scarce [14]. However, a limited number of molecularly identified strains suggest association of this yeast with plants [14]. Interestingly, C. wieringae was more abundant than C. magnus in our study and according to these results, C. wieringae appeared to be an important component of the phylloplane community, probably overlooked and accounted as C. albidus due to the use of less accurate phenotypic identification approaches in previous studies, e.g. [17, 34].
Molecular reassessment of the strains identified as C. laurentii placed most of the studied isolates within the victoriae clade (Tremellales, Tremellomycetes), with C. victoriae being the most frequently found species (Table 1; Fig. 2). In our study, the occurrence of this species was restricted to the plant material, which contrasts with previous reports where C. victoriae was also isolated from soil substrates [9, 21, 52, 56]. A few reports also describe the isolation of C. victoriae from apples, flower nectar and leaves of Mediterranean plants [14]. Our results additionally support phylloplane-related origin of this yeast, which was found in both studied regions and from various plants (Table 1; Table S1). We suggest that a large part of previous reports on C. laurentii isolates from plant surfaces [2, 17, 19] probably refer to C. victoriae. According to the origin of strains identified with molecular methods (this study; [14]), C. victoriae seems to have a wide distribution range. Although global distribution patterns of microbial species are currently debatable (e.g. [48]), C. victoriae might represent a good example of a globally spread phylogenetic species. According to our results, this species is extremely polymorphic. It displays interspecific variability of morphological and physiological characteristics as well as considerable genetic variability (see also [9]). Studies utilizing additional genetic markers are required to answer the question whether C. victoriae in its current circumscription is indeed widespread or it represents a complex of cryptic species.
About 20 % of isolates previously assigned to C. laurentii were identified as C. tephrensis in the present study. Although this species was originally isolated from soil [51], and latter isolated from this substrate by other authors [21, 52], the probable soil origin of C. tephrensis is controversial. An extensive study of different soils in Germany reported C. tephrensis as being a transient species in these substrates, due to its rare occurrence and low abundance [62]. In other example, C. tephrensis was reported in a large-scale study on soil yeasts but with a very low frequency, except for Iceland soils [52]. Thus, the ecology of this species is still unclear (see also [14]). However, evidence for association of C. tephrensis with plant material is provided in this study (Table 1). Additionally, several CBS collection strains (CBS 8993, CBS 9012, CBS 9023 and CBS 9799) representing this species were also isolated from plants (e.g. [20]). We suggest that C. tephrensis may be associated with the phylloplane, from where it can reach neighbour substrates, like soil.
Yeasts identified as C. diffluens have been repeatedly observed in dry habitats, like deserts [2, 7], seasonally dried peat and house dust ([18] and references therein). Bab’eva and Chernov [2] suggested that the occurrence of C. diffluens may indicate a low water content in the environment. We found that isolates identified as C. diffluens using MSR approaches were mostly conspecific, but belong to a phylogenetically distinct species, C. albidus (Fig. 3). Remarkably, two reference strains of phenotypic C. diffluens (KBP 3824 and 3826) isolated from sub-tropical deserts by Chernov et al. [7] were also identified as C. albidus (Table 1; Fig. 3). The presence of C. albidus in dry habitats agrees with results of previous experiments, which demonstrated a good survival of these yeasts in sandy soil due to the abundant production of polysaccharide capsules [50]. Interestingly, the assessment of soil yeast distribution along a large latitudinal gradient using molecular identification tools showed that C. albidus occurrence was negatively correlated with the rainfall [52]. We suggest that a large proportion of previous reports describing the isolation of C. diffluens [2, 7, 18, 34] may in fact refer to C. albidus.
Interestingly, the identification of the soil-borne yeasts C. terricola and C. podzolicus using conventional physiological criteria was consistent with the identification using molecular tools. Recently, Yurkov et al. [61] demonstrated that yeast communities in forest soils largely consist of distantly related species, which are likely to be successfully identified using physiological tests. Unlike under-sampling, the presence of cryptic species does not cause significant underestimation of soil yeast diversity [61]. In the present study, we found soils to be inhabited by a few number of yeasts, C. albidus, C. terricola and C. podzolicus (Table 1). In contrast, plant substrates harbour diverse physiologically indistinguishable yeast species, such as C. victoriae, C. carnescens, C. heimaeyensis, C. tephrensis and C. foliicola. The occurrence of cryptic species seem to pose a real problem for assessing the biodiversity in the phylloplane when using only MSR approaches to identify yeast isolates.
Red yeasts were repeatedly reported as dominant phylloplane inhabitants and the two species R. babjevae and S. roseus were commonly isolated from plant material worldwide, e.g. [2, 12, 25, 34]. In contrast, the association of the other two Rhodotorula species isolated in this work, R. pinicola and R. slooffiae, with any habitat is currently unclear. While R. slooffiae is a rather frequent species [39], R. pinicola was known from a few isolates only. Our results suggest that R. pinicola might regularly inhabit plant substrates and have a wide distribution range (Table 1). Furthermore, this species might be mislabelled and accounted as R. minuta in earlier studies. However, it should be stressed out that additional studies are required to reassess distribution of red-coloured yeasts, as they are often difficult to differentiate using assimilation tests, e.g. [39].
Although the precise habitat of Cu. cygneicollum (R. fujisanensis) remains unclear, the majority of known strains were isolated from different types of plant material [25, 34, 40]. Our results support this observation as we found this yeast across all stages of plants decomposition, from live parts to litter and the topsoil (Table 1). Several anamorphic non-pigmented strains originally identified as Rhodotorula spp. were assigned to L. scottii. This species occupies diverse habitats in the temperate zone and were mostly reported from soils and water habitats [41]. To our knowledge, high abundance of this yeast in the phylloplane of vascular plants has not been reported before.
Species Recognition Approaches
Phylogenetic species boundaries do not necessarily coincide with the species boundaries based on morphological or biochemical characters or reproductive isolation, e.g. [48]. Despite their current popularity, biodiversity assessments using species recognition solely based on phylogenetic approaches are known to be sensitive to the sampling effort [45, 53, 61], since PSR is able to resolve smaller entities with more restricted geographic ranges and decreased abundances [1]. Re-identification of Cryptococcus yeasts performed with molecular tools demonstrated that taxonomical units determined with MSR correspond mostly to the level of phylogenetic clades or roughly genera when considering polyphyly of this genus [14]. Even though MSR and PSR seem to result in entities corresponding to two different taxonomic levels in yeast species, linking “phenotypic” and phylogenetic species is hardly feasible due to widespread mislabelling of polymorphic species identified with MSR as C. laurentii and C. albidus. Even though our results showed the majority of PSR-based species identified in MSR-based C. laurentii isolates were mostly placed within a monophyletic victoriae clade in Tremellales (Table 1; Fig. 1), this clade is distantly related to the Bulleromyces clade that includes the type strain of C. laurentii [14, 59]. The use of conventional phenotypic methods alone in biodiversity studies is feasible to some extent only. Our results suggest that these methods could provide reliable resolution at the level of phylogenetic clades and can be utilized for the analyses of functional traits in yeasts.
Conclusions
Appropriate re-identification of species previously resolved with phenotypic characters enabled comparisons with previous surveys that also used molecular methods and revealed frequent occurrence and wide distribution of C. wieringae, C. tephrensis and R. pinicola, species known previously from only a few isolates, and C. victoriae and C. albidus being long reported under incorrect names due to the inaccuracy of phenotypic identification. Comparison with previous studies confirms certain trends and reveals others that clearly deserve further studies, including the definition of yeast characteristics that determine their vertical (substrates) and horizontal (spatial) distribution. Even though our study revealed some congruence between distribution patterns of yeasts identified with MSR and PSR approaches, strain profiling using PCR fingerprinting techniques suggests a rather complex interspecific structure of many basidiomycetous yeast species that should be addressed in further studies.
Molecular typing may additionally help to uncover interspecific heterogeneity and population structure and, thus, reveal possible correlations between genotype and geographical origin or other ecologically relevant parameters. Geographical population structure and allopatric speciation, previously observed in ascomycetous fungi, appears to be present to some extent among basidiomycetes as well.
References
Agapow PM, Bininda-Emonds OR, Crandall KA, Gittleman JL, Mace GM, Marshall JC, Purvis A (2004) The impact of species concept on biodiversity studies. Q Rev Biol 79:161–179
Bab’eva IP, Chernov IY (1995) Geographical aspects of yeast ecology. Physiol Gen Biol Rev 9:1–54
Bab’eva IP, Reshetova IS (1975) A new yeast species Candida podzolica sp. nov. isolated from the soil. Microbiologia 44:333–338 English abstract
Barnett JA, Payne RW, Yarrow D (1990) Yeasts: characteristics and identification, 2nd edn. Cambridge University Press, Cambridge
Birkhofer K, Schöning I, Alt F, Herold N, Klarner B et al (2012) General relationships between abiotic soil properties and soil biota across spatial scales and different land-use types. PLoS ONE 7:e43292
Chernov IY (2005) Latitude-zonal and spatial-successional trends in yeast fungi distribution. Zh Obshch Biol 66:123–135 English abstract
Chernov IY, Bab’eva IP, Reshetova IS (1997) Synecology of yeast fungi in subtropical deserts. Usp Sovrem Biol 117:584–602 English abstract
Coelho MA, Gonçalves P, Sampaio JP (2011) Evidence for maintenance of sex determinants but not of sexual stages in red yeasts, a group of early diverged basidiomycetes. BMC Evol Biol 11:249
de Garcia V, Zalar P, Brizzio S, Gunde-Cimerman N, van Broock M (2012) Cryptococcus species (Tremellales) from glacial biomes in the southern (Patagonia) and northern (Svalbard) hemispheres. FEMS Microbiol Ecol 82:523–539
Dunlap CA, Evans KO, Theelen B, Boekhout T, Schisler DA (2007) Osmotic shock tolerance and membrane fluidity of cold-adapted Cryptococcus flavescens OH 182.9, previously reported as C. nodaensis, a biocontrol agent of Fusarium head blight. FEMS Yeast Res 7:449–458
Fonseca Á (1992) Utilization of tartaric acid and related compounds by yeasts: taxonomic implications. Can J Microbiol 38:1242–1251
Fonseca Á, Inácio J (2006) Phylloplane Yeasts. In: Peter G, Rosa C (eds) Biodiversity and Ecophysiology of Yeasts. Springer-Verlag, Berlin, pp 263–301
Fonseca Á, Scorzetti G, Fell JW (2000) Diversity in the yeast Cryptococcus albidus and related species as revealed by ribosomal DNA sequence analysis. Can J Microbiol 46:7–27
Fonseca Á, Boekhout T, Fell JW (2011) Cryptococcus Vuillemin. In: Kurtzman CP, Fell JW, Boekhout T (eds) The yeasts, a taxonomic study, 5th edn. Elsevier, Amsterdam, pp 1661–1737
Gadanho M, Sampaio JP (2002) Polyphasic taxonomy of the basidiomycetous yeast genus Rhodotorula: Rh. glutinis sensu stricto and Rh. dairenensis comb. nov. FEMS Yeast Res 2:47–58
Ganter PF, Cardinali G, Giammaria M, Quarles B (2004) Correlations among measures of phenotypic and genetic variation within an oligotrophic asexual yeast, Candida sonorensis, collected from Opuntia. FEMS Yeast Res 4:527–540
Glushakova AM, Chernov IY (2004) Seasonal dynamics in a yeast population on leaves of the common wood sorrel Oxalis acetosella L. Microbiology 73:184–188
Glushakova AM, Zheltikova TM, Chernov IY (2004) Groups and sources of yeasts in house dust. Microbiology 73:94–98
Golubtsova YV, Glushakova AM, Chernov IY (2007) The seasonal dynamics of yeast communities in the rhizosphere of soddy-podzolic soils. Eurasian Soil Sci 40:875–879
Herzberg M, Fischer R, Titze A (2002) Conflicting results obtained by RAPD-PCR and large-subunit rDNA sequences in determining and comparing yeast strains isolated from flowers: a comparison of two methods. Int J Syst Evol Micr 52:1423–1433
Hong SG, Lee KH, Bae KS (2002) Diversity of yeasts associated with natural environments in Korea. Journal Microbiol 40:55–62
Inácio J, Rodrigues MG, Sobral P, Fonseca Á (2004) Characterisation and classification of phylloplane yeasts from Portugal related to the genus Taphrina and description of five novel Lalaria species. FEMS Yeast Res 4:541–555
Inácio J, Portugal L, Spencer-Martins I, Fonseca Á (2005) Phylloplane yeasts from Portugal: seven novel anamorphic species in the Tremellales lineage of the Hymenomycetes (Basidiomycota) producing orange-coloured colonies. FEMS Yeast Res 5:1167–1183
Kachalkin AV, Yurkov AM (2012) Yeast communities in Sphagnum phyllosphere along the temperature-moisture ecocline in the boreal forest-swamp ecosystem and description of Candida sphagnicola sp. nov. Anton Leeuw Int J G 102:29–43
Kachalkin AV, Glushakova AM, Yurkov AM, Chernov IY (2008) Characterization of yeast groupings in the phyllosphere of Sphagnum mosses. Microbiology 77:474–481
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform (describes the FFT-NS-1, FFT-NS-2 and FFT-NS-i strategies). Nucleic Acids Res 30:3059–3066
Kurtzman CP, Fell JW (1998) The yeasts, a taxonomic study, 4th edn. Elsevier, Amsterdam
Kurtzman CP, Fell JW (2006) Yeast systematics and phylogeny—implications of molecular identification methods for studies in ecology. In: Peter G, Rosa C (eds) Biodiversity and ecophysiology of yeasts. Springer-Verlag, Berlin, pp 11–30
Lachance M-A (2006) Yeast biodiversity: how many and how much. In: Peter G, Rosa C (eds) Biodiversity and ecophysiology of yeasts. Springer-Verlag, Berlin, pp 1–11
Lachance M-A, Starmer WT, Rosa CA, Bowles JM, Barker JSF, Janzen DH (2001) Biogeography of the yeasts of ephemeral flowers and their insects. FEMS Yeast Res 1:1–8
Lachance M-A, Dobson J, Wijayanayaka DN, Smith AM (2010) The use of parsimony network analysis for the formal delineation of phylogenetic species of yeasts: Candida apicola, Candida azyma, and Candida parazyma sp. nov., cosmopolitan yeasts associated with floricolous insects. Anton Leeuw Int J G 97:155–170
Leducq JB, Charron G, Samani P, Dubé AK, Sylvester K et al (2014) Local climatic adaptation in a widespread microorganism. P Roy Soc B-Biol Sci 281:20132472
Liti G, Carter DM, Moses AM, Warringer J, Parts L et al (2009) Population genomics of domestic and wild yeasts. Nature 458:337–341
Maksimova IA, Chernov IY (2004) Community structure of yeast fungi in forest biogeocenoses. Microbiology 73:474–481
Maksimova IA, Yurkov AM, Chernov IY (2009) Spatial structure of epiphytic yeast communities on fruits of Sorbus aucuparia L. Biol Bull 36:613–618
Naumov GI, Naumova ES, Molina FI (2000) Genetic variations among European strains of Saccharomyces paradoxus: results from DNA fingerprinting. Syst Appl Microbiol 23:86–92
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Sampaio JP (1999) Utilization of low molecular weight aromatic compounds by heterobasidiomycetous yeasts: taxonomic implications. Can J Microbiol 45:491–512
Sampaio JP (2011) Rhodotorula Harrison (1928). In: Kurtzman CP, Fell JW, Boekhout T (eds) The yeasts, a taxonomic study, 5th edn. Elsevier, San Diego, pp 1873–1928
Sampaio JP (2011) Curvibasidium Sampaio and Golubev (2004). In: Kurtzman CP, Fell JW, Boekhout T (eds) The yeasts, a taxonomic study, 5th edn. Elsevier, San Diego, pp 1413–1418
Sampaio JP (2011) Leucosporidium Fell, Statzell, Hunter & Phaff (1969). In: Kurtzman CP, Fell JW, Boekhout T (eds) The yeasts, a taxonomic study, 5th edn. Elsevier, San Diego, pp 1489–1494
Sampaio JP, Gadanho M, Santos S, Duarte FL, Pais C, Fonseca Á, Fell JW (2001) Polyphasic taxonomy of the basidiomycetous yeast genus Rhodosporidium: R. kratochvilovae and related anamorphic species. Int J Syst Evol Micr 51:687–697
Sampaio JP, Golubev WI, Fell JW, Gadanho M, Golubev NW (2004) Curvibasidium cygneicollum gen. nov., sp. nov., Curvibasidium pallidicorallinum sp. nov., novel taxa in the Microbotryomycetidae, Urediniomycetes, and the relationship with Rhodotorula fujisanensis and Rhodotorula nothofagi. Int J Syst Evol Micr 54:1401–1407
Scorzetti G, Fell JW, Fonseca Á, Statzell-Tallman A (2002) Systematics of basidiomycetous yeasts: a comparison of large subunit D1/D2 and internal transcribed spacer rDNA regions. FEMS Yeast Res 2:495–517
Sites JW Jr, Crandall KA (1997) Testing species boundaries in biodiversity studies. Conserv Biol 11:1289–1297
Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML web-servers. Syst Biol 75:758–771
Starmer WT, Aberdeen V, Lachance M-A (2006) The biogeographic diversity of cactophilic yeasts. In: Peter G, Rosa C (eds) Biodiversity and ecophysiology of yeasts. Springer-Verlag, Berlin, pp 485–499
Taylor JW, Turner E, Townsend JP, Dettman JR, Jacobson D (2006) Eukaryotic microbes, species recognition and the geographic limits of species: examples from the kingdom Fungi. Philos T R Soc B 361:1947–1963
Thomas-Hall S, Watson K, Scorzetti G (2002) Cryptococcus statzelliae sp. nov. and three novel strains of Cryptococcus victoriae, yeasts isolated from Antarctic soils. Int J Syst Evol Micr 52:2303–2308
Vishniac HS (1995) Simulated in situ competitive ability and survival of a representative soil yeast, Cryptococcus albidus. Microb Ecol 30:309–320
Vishniac HS (2002) Cryptococcus tephrensis sp. nov., and Cryptococcus heimaeyensis sp. nov.; new anamorphic basidiomycetous yeast species from Iceland. Can J Microbiol 48:463–467
Vishniac HS (2006) A multivariate analysis of soil yeasts isolated from a latitudinal gradient. Microbl Ecol 52:90–103
Walsh PD (2000) Sample size for the diagnosis of conservation units. Conserv Biol 14:1533–1537
Wang QM, Boekhout T, Bai FY (2011) Cryptococcus foliicola sp. nov. and Cryptococcus taibaiensis sp. nov., novel basidiomycetous yeast species from plant leaves. J Gen Appl Microbiol 57:285–291
Whitaker RJ (2006) Allopatric origins of microbial species. Philos T R Soc B 361:1975–1984
Wuczkowski M, Prillinger H (2004) Molecular identification of yeasts from soils of the alluvial forest national park along the river Danube downstream of Vienna, Austria (“Nationalpark Donauauen”). Microbiol Res 159:263–275
Yarrow D (1998) Methods for the isolation, maintenance and identification of yeasts. In: Kurtzman CP, Fell JW (eds) The yeasts, a taxonomic study, 4th edn. Elsevier, Amsterdam, pp 77–100
Yurkov AM, Chernov IY (2005) Geographical races of certain species of ascomycetous yeasts in the Moscow and Novosibirsk regions. Microbiology 74:597–601
Yurkov AM, Golubev WI (2013) Phylogenetic study of Cryptococcus laurentii mycocinogenic strains. Mycol Prog 12:777–782
Yurkov AM, Maksimova IA, Chernov IY (2004) The comparative analysis of yeast communities structure in birch forests of European Russia and Western Siberia. Mikol Fitopatol 38:71–79 English abstract
Yurkov AM, Kemler M, Begerow D (2011) Species accumulation curves and incidence-based species richness estimators to appraise the diversity of cultivable yeasts from beech forest soils. PLoS ONE 6:e23671
Yurkov AM, Kemler M, Begerow D (2012) Assessment of yeast diversity in soils under different management regimes. Fungal Ecol 5:24–35
Yurkov AM, Krüger D, Begerow D, Arnold N, Tarkka MT (2012) Basidiomycetous yeasts from Boletales fruiting bodies and their interactions with the mycoparasite Sepedonium chrysospermum and the host fungus Paxillus. Microb Ecol 63:295–303
Acknowledgments
AY received funds from the cooperation agreement “Acordo Cultural Portugal - Rússia” (GRICES), Russian Fund for Basic Research (RFBR 04-04-48713) and Fundação para a Ciência e a Tecnologia (Grants SFRH/BPD/79620/2011, PTDC/BIA-BIC/4585/2012). J.P. Sampaio and M. Gadanho (CREM, Portugal) are acknowledged for obtaining sequence data from two isolates. A.M. Glushakova (MSU, Moscow) and A.A. Sakharov assisted in samples collection.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yurkov, A., Inácio, J., Chernov, I.Y. et al. Yeast Biogeography and the Effects of Species Recognition Approaches: The Case Study of Widespread Basidiomycetous Species from Birch Forests in Russia. Curr Microbiol 70, 587–601 (2015). https://doi.org/10.1007/s00284-014-0755-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-014-0755-9