
Abstract
Corynebacterium pseudotuberculosis is the etiologic agent of caseous lymphadenitis a chronic infectious disease affecting small ruminants. The 2D-DIGE technique was used to compare the exoproteomes of two C. pseudotuberculosis biovar ovis strains isolated from goat (strain 1002) and sheep (strain C231). Seventeen proteins differentially produced were identified here. Nine proteins appeared over-produced in the exoproteome of 1002 goat strain and 8 in that of C231 sheep strain. These proteins were related to various biological functions, such as the cell envelope, respiratory metabolism and proteolysis. This proteomic analysis revealed strain-specific exoproteins although each of the corresponding genes was found in both strain genomes. Such differential expression pattern may reflect inter-strain differences in adaptation to a specific host, in pathogenicity and or in antigenicity of this pathogenic bacterium.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Corynebacterium pseudotuberculosis is a Gram-positive, facultative intracellular pathogen, and the etiologic agent of caseous lymphadenitis (CLA), a chronic infectious disease affecting small ruminants [9]. Despite a huge impact of CLA in small ruminant herds, C. pseudotuberculosis is still poorly documented and our understanding of its pathogenesis is partial. A few virulence factors of C. pseudotuberculosis were identified in previous works, including phospholipase D [15], toxic cell wall lipids [12], iron transporters belonging to the ABC proteins family [5] and a serine protease [32].
Comparative proteomics has been used to identify virulence factors and to gain further information about the physiology of various pathogens such as Listeria monocytogenes [29], Staphylococcus aureus [17] or Clostridium perfringens [27]. These studies took account of secreted/extracellular proteins, a protein fraction which indeed contains factors involved in adhesion and invasion of the host cell, and in survival, persistence and intracellular proliferation [29].
We recently reported the use of a gel-free proteomic approach to compare the exoproteomes of two C. pseudotuberculosis strains 1002 and C231 that both belong to the biovar ovis, but were isolated from different hosts: goat and sheep, respectively. This study enabled detection of qualitative and quantitative changes between the exoproteome of the two strains. Proteins identified were associated with bacterial physiology and included putative virulence factors [22]. However, such approach only gives a partial view of the protein sample contents. More sensitive techniques, such as the two-dimensional differential in-gel electrophoresis (2D-DIGE), have been described and allow to achieve a more complete analysis of proteomes. This multiplex technique allows analyzing differences between protein samples resolved on the same gel, in the presence of an internal standard, enabling more accurate results. 2D-DIGE demonstrated advantages over the conventional 2-DE in quantitative studies [1]. Here, 2D-DIGE was used to identify quantitative changes in the exoproteomes of C. pseudotuberculosis 1002 and C231.
Materials and Methods
Bacterial Strains and Culture Conditions
Corynebacterium pseudotuberculosis biovar ovis strain 1002, isolated from a caprine host in Brazil, and C. pseudotuberculosis biovar ovis strain C231, isolated from an ovine host in Australia, were maintained in brain–heart-infusion broth (BHI—HiMedia Laboratories Pvt. Ltd., India) at 37 °C. For proteomic analysis, both strains were cultivated in chemically defined medium (CDM) [(Na2HPO4·7H2O (12.93 g/L), KH2PO4 (2.55 g/L), NH4Cl (1 g/L), MgSO4·7H2O (0.20 g/L), CaCl2 (0.02 g/L) and 0.05 % (v/v) Tween 80]; 4 % (v/v) MEM Vitamins Solution 100× (Invitrogen, Gaithersburg, MD, USA); 1 % (v/v) MEM Amino Acids Solution 50× (Invitrogen); 1 % (v/v) MEM Non-Essential Amino Acids Solution 100× (Invitrogen); and 1.2 % (w/v) filter-sterilized glucose) [19].
Three-Phase Partitioning
The three-phase partitioning protocol (TPP) was used to extract extracellular proteins [24]. Briefly, each strain was cultivated in triplicate on CDM and after reaching late exponential growth (DO600nm = 1.3). Bacterial cells were pelleted by centrifugation for 20 min at 2,700×g, and culture supernatants were filtered using 0.22 μm filters. Ammonium sulfate 6 g/L was added to supernatant samples, and the pH was set to 4.0. N-butanol 20 mL/L was then added to each 20 mL of sample, left 1 h at room temperature and centrifuged for 10 min at 1,350×g at 4 °C. The interfacial precipitate was re-suspended in 1 mL Tris–HCl (20 mM, pH 7.2) + 10 μL Protease Inhibitor Mix (GE Healthcare, Piscataway, NJ, USA).
2D-DIGE
For 2D-DIGE experiment, the protein samples (50 μg) of each strain were labeled separately with 400 pmol of either Cy3 or Cy5 CyDye DIGE Fluor minimal dyes (GE Healthcare, Piscataway, NJ, USA), and an internal standard was prepared using 25 μg of each bacterial protein extract (Cp1002 + CpC231) and labeled with 400 pmol of Cy2 (GE Healthcare). The labeled samples were then mixed and combined with a sample buffer containing 7 M urea, 2 M thiourea, 4 % CHAPS, 0.2 % DTT and 0.002 % bromophenol blue and applied to 18 cm pH 3–10 NL strips (GE Healthcare). Isoelectric focusing was performed using the IPGphor 2 (GE Healthcare) for a total of 60,000 Vh. The strips were then brought to equilibrium for 15 min in 10 mL of equilibration buffer I (Tris–HCl 50 mM pH 8.8, urea 6 M, glycerol 30 %, SDS 2 %, bromophenol blue 0.002 %, 100 mg dithiothreitol) followed by 15 min in 10 mL of equilibration buffer II (Tris–HCl 50 mM pH 8.8, urea 6 M, glycerol 30 %, SDS 2 %, bromophenol blue 0.002 %, iodoacetamide 250 mg). After equilibration, proteins were separated in 12 % acrylamide/bis-acrylamide gels with an Ettan DaltSix II system (GE Healthcare), and the gels were then scanned between low fluorescence glass plates at a 50 nm resolution. The three fluorophores were imaged at excitation wavelengths of Cy3/580 nm, Cy5/670 nm and Cy2/520 nm, and gel images were cropped and analyzed using the Image Master 2D Platinum 7.0 DIGE Software (GE Healthcare). Spots with at least a 2.0-fold volume ratio change and ANOVA t test P value less than 0.01 were selected for identification.
In-Gel Trypsin Digestion, Mass Spectrometry and Protein Identification
Protein spots were excised from the gels using an Ettan Spot Picker (GE Healthcare) and in-gel digestion was carried out using trypsin enzyme (Promega, Sequencing Grade Modified Trypsin, Madison, WI, USA). The peptides were then concentrated to a volume of 10 μL using a speed vacuum, desalinated and concentrated using ZIP-TIP C18 tips (Eppendorf, Hamburg, Germany). The samples were subsequently analyzed by MS and MS/MS modes, using a MALDI-TOF/TOF mass spectrometer Autoflex III™ (Bruker Daltonics, Billerica USA). The equipment was controlled in a positive/reflector way using the FlexControlTM software (Brucker Daltonics), and calibration was performed using standard peptide samples (angiotensin II, angiotensin I, substance P, bombesin, ACTH clip 1–17, ACTH clip 18–39, somatostatin 28, bradykinin Fragment 1–7, renin Substrate tetradecapeptide porcine) (Bruker Daltonics). The peptides were added to the alpha-cyano-4-hydroxycinnamic acid matrix, applied on an AnchorChipTM 600 plate (Brucker Daltonics) and analyzed by Autoflex III. The search parameters were as follows: peptide mass fingerprint; enzyme; trypsin; fixed modification, carbamido methylation (Cys); variable modifications, oxidation (Met); mass values, monoisotopic; maximum missed cleavages, 1; and peptide mass tolerance of 0.05 % Da (50 ppm). The results obtained by MS/MS were used to identify proteins utilizing the MASCOT® (http://www.matrixscience.com) program and compared to NCBI databases.
Bioinformatics Analysis
The Blast2GO program was used to classify the protein’s functionality [8]. To predict the sub-cellular localization of proteins, we used the following programs: SurfG+v1.0 [2], SecretomeP v2.0 [3] and TatP v1.0 [4].
Results and Discussion
2D-DIGE and Mass Spectrometry
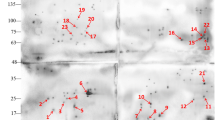
In this study, the 2D-DIGE was used for a quantitative analysis and a comparison of the exoproteomes of C. pseudotuberculosis 1002 and C231, isolated from of goat and sheep, respectively (Fig. 1). Using this technique, 18 spots were found differentially produced, selected and subjected to mass spectrometry analysis. Nine out of these 18 proteins were specifically found over-produced in strain 1002 supernatants (Supplementary file 1 and Fig. 2) and 8 were over-produced in strain C231 supernatants (Supplementary file 2 and Fig. 2 ). Of note, the genes corresponding to each of these proteins are found in both 1002 and C231 genomes suggesting that the differences observed at the proteome level are due to strain-specific abilities in expressing these particular genes, rather than differences in gene content.

Comparative display of differential in-gel electrophoresis (DIGE) of the extracellular fractions of strains 1002 and C231 of C. pseudotuberculosis. Overlay of the three color images Cy3 (colored green), Cy5 (colored red) and Cy2 (colored blue) derived from a single gel

Differential expression pattern of the proteins spots. Expanded views of the proteins spots differentially produced, between the strains 1002 and C231, identified by MS/MS
Interestingly, various selected spots actually contained the same unique protein. For example, trehalose corynomycolyl transferase C was found at the expected molecular mass in C231 (spot 53; Supplementary file 2) and at a lower observed mass in 1002 (spot 87; Supplementary file 1), suggesting a proteolytic cleavage occurred in strain 1002. Cytochrome c oxidase sub-unit 2 was found over-produced in C231 in two different spots, at the expected size but significantly lower pI (spot 147; Supplementary file 2) and at an observed mass slightly higher than the theoretical mass (spot 125; Supplementary file 2). These observations suggest the occurrence of post-translational modifications, which are commonly detected in studies based in gel-dependent system [25]. Such post-translational modifications were shown to regulate several cellular functions, for example, in S. aureus, post-translational modifications (phosphorylation) play a key role in pathogenesis, through modulation of adhesion to and invasion of host cells [21]. The exact nature of the putative post-traductional modifications observed here is still to be determined. Whether and how such modifications can affect pathogenesis and or host specificity in C. pseudotuberculosis remains unknown.
Predicting the Sub-Cellular Localization of Identified Exoproteins
In silico predictions of sub-cellular localization of C. pseudotuberculosis, extracellular proteins were performed using the software SurfG+ [2]. The predictions point out the presence of 13 secreted proteins, 2 proteins possibly associated with the cell wall and 1 cytoplasmic protein. The protein predicted as cytoplasmatic was glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (spot 101, Supplementary file 1); this protein is associated with the lipid biosynthesis, carbohydrate metabolis and oxireduction process. However, despite being predicted as a cytoplasmic protein, several studies have detected GAPDH in different sub-cellular locations (membrane, cell surface and extracellular) and developing different functions; due to these features, this protein is classified as moonlighting proteins; these proteins show distinct functional behavior depending on the cell type, cellular localization and multiple binding sites [14]. Several of the moonlighting functions of GAPDH have been studied in different Gram-positive and Gram-negative pathogens, showing that this protein plays important role during the adhesion and invasion process to host cell [11, 18, 23]. The functional prediction of GPDH by Blast2GO in C. pseudotuberculosis shows the presence of two distinct functions: (1) oxireduction activity and (2) binding nucleotide showed the moonlight behavior this protein in this pathogen (Supplementary file 1). However, further studies are needed to uncover the true moonlighting role of GPDH in C. pseudotuberculosis.
Functional Classification of Differentially Produced Proteins
The differentially produced proteins identified here were related to various physiological functions. Two proteins (spots 204 and 250) found in strain C231, and one protein (spot 149) found in 1002 were of unknown function when submitted to a Blast2GO search [8]. Their putative role in the physiology and/or virulence of C. pseudotuberculosis thus remains to be determined. Four proteins detected in 1002 strain (spot 28, 66, 85 and 86, Supplementary file 1) and one protein detected in C231 strain (spot 199, Supplementary file 2) are associated with cell envelope and have a hydrolase activity. Such hydrolases partially digest the cell wall during the bacterial growth and thus participate in the turnover of peptidoglycan during the cell growth, in cell division or in cell autolysis [31]. Peptidoglycan is essential for cell viability and shape. It also controls the internal osmotic pressure [13]. Among, the detected proteins with hydrolases activity, spot 28 (Supplementary file 1) was identified as a neuraminidase in 1002 supernatants. This protein acts in the hydrolysis process of glycoproteins by cleaving sialic acid residues. Moreover, it was shown to be involved in the virulence of pathogens like Pseudomonas aeruginosa and Streptococcus pneumoniae, by promoting cell adhesion and cell invasion [7, 30].
Other proteins are found differentially produced and are also associated with cell envelope: Mycolyltransferases (spot 87, 178, Supplementary file 1; spot 53, 123, Supplementary file 2) were previously shown to be associated with the cell envelope in Mycobacterium tuberculosis. This class of proteins is involved in the biosynthesis of corynemycolyl components that are associated with the envelope structure. These components are formed from high molecular weight chain fatty acids and are the largest cell wall constituents of Mycobacterium, Nocardia and Corynebacterium [28]. Changes in the quantity and structure of these fatty acids can affect the permeability, fluidity and other physical characteristics of the bilayer membrane of these bacteria and consequently may influence bacterial growth [33]. Furthermore, these constituents of the cell envelope also play important roles in the mechanisms of pathogenicity, due to their ability to form an impermeable asymmetric lipid bilayer that contributes to the resistance and survival of the bacteria in the hostile environment of macrophages [10].
A hypothetical protein (spot 225, Supplementary file 1) showed conserved domains the calcium ion binding. Calcium and calcium-binding proteins are involved in numerous bacterial processes, such as chemotaxis, sporulation, virulence, molecules transport, phosphorylation, septation and stability of the cell envelope [20]. Several studies showed that calcium also influences the formation of biofilms. Thickness of P. aeruginosa biofilm increase in the presence of CaCl2 compared to growth in medium without CaCl2 supplementation [26]. The formation of biofilm by V. cholerae is dependent upon calcium and biofilms dissolve when the medium is depleted of calcium [16].
Bacteria that show characteristics of aerobic or anaerobic facultative growth, such as C. pseudotuberculosis, need oxygen as exogenous electron acceptor for respiration. The Cytochrome c oxidase sub-unit 2 (spot 125, 147, Supplementary file 2) is involved in this process [6], and was detected in this work.
Exploring C. pseudotuberculosis Exoproteome by Different Proteomics Approaches
The results obtained here were compared to those obtained by Pacheco et al. [22] in a previous study. Most of the differentially produced proteins identified in this work were shown over-produced as well in Pacheco et al. [22]. However, this study revealed some discrepancies with this previous study. Proteins ADL20134, ADL20574 and ADL21814 were indeed found over-produced in 1002 here (Table 1), whereas they were previously reported over-produced in C231 [22] and vice versa for proteins ADL11400 and ADL10895. We also identified some additional proteins, for example, lysozyme M1 (spot 86, Supplementary file 1), GPDH (spot 101, Supplementary file 1) and hypothetical protein (spot 250, Supplementary file 2) that were not previously detected. Such differences may be linked to the experimental procedure used: these differentially produced or additional proteins may indeed be revealed in samples prepared, in this work, from cultures in late exponential growth phase whereas they did not appear in Pacheco et al. [22] where proteins were prepared from cultures in early exponential phase. On the other hand, sensitivity of the 2D-DIGE technique (as compared to the gel-free approach adopted in Pacheco et al. [22].) might also explain the additional proteins that were identified here. These results demonstrated that comparative analysis, combining different proteomic approaches, is a powerful strategy to characterize a proteome.
Conclusions
Exoproteome analysis of C. pseudotuberculosis strains 1002 and C231 revealed differential production patterns, which may be related to differences in host adaptation, pathogenicity or antigenicity of this pathogen. The genes corresponding to the differentially produced proteins are present in both strains, suggesting differences related to the ability of each strain to express these genes in these growth conditions. The results obtained here complement previous comparative genomic and proteomic studies, adding data regarding the biology and virulence of C. pseudotuberculosis. Investigating the proteomes of C. pseudotuberculosis strains isolated from various hosts will help understand how bacteria are able to adapt to specific hosts, and provide excellent candidates for targeted studies of the molecular basis of C. pseudotuberculosis pathogenesis in small ruminant.
References
Alban A, David SO, Bjorkesten L, Andersson C, Sloge E, Lewis S, Currie I (2003) A novel experimental design for comparative two-dimensional gel analysis: two-dimensional difference gel electrophoresis incorporating a pooled internal standard. Proteomics 3:36–44
Barinov A, Loux V, Hammani A, Nicolas P, Langella P, Maquin E, van de Guchte M (2009) Prediction of surface exposed proteins in Streptococcus pyogenes, with a potential application to other gram-positive bacteria. Proteomics 9:61–73
Bendtsen JD, Kiemer L, Fausboll A, Brunak S (2005) Non-classical protein secretion in bacteria. BMC Microbiol 5:58
Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S (2005) Prediction of twin-arginine signal peptides. BMC Bioinformatics 6:167
Billington SJ, Esmay PA, Songer JG, Jost BH (2002) Identification and role in virulence of putative iron acquisition genes from Corynebacterium pseudotuberculosis. J Bacteriol 180:3233–3236
Bott M, Niebisch A (2003) The respiratory chain of Corynebacterium glutamicum. J Biotechnol 4:129–153
Cacalano G, Kays M, Saiman L, Prince A (1992) Production of the Pseudomonas aeruginosa neuraminidase is increased under hyperosmolar conditions and is regulated by genes involved in alginate expression. J Clin Invest 89:1866–1874
Conesa A, Gotz S, Garica-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Dorella FA, Pacheco LG, Oliveira SC, Miyoshi A, Azevedo V (2006) Corynebacterium pseudotuberculosis: microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Vet Res 37:201–218
Dubnau E, Chan J, Raynaud C, Mohan VP, Lanéelle MA, Yu K, Quémard A, Smith I, Daffé M (2000) Oxygenated mycolic acids are necessary for virulence of Mycobacterium tuberculosis in mice. Mol Microbiol 36:630–637
Egea L, Aguilera L, Giménez R, Sorolla MA, Aguilar J, Badía J, Baldoma L (2007) Role of secreted glyceraldehyde-3-phosphate dehydrogenase in the infection mechanism of enterohemorrhagic and enteropathogenic Escherichia coli: interaction of the extracellular enzyme with human plasminogen and fibrinogen. Int J Biochem Cell Biol 39:1190–1203
Hard GC (1975) Comparative toxic effect on the surface lipid of Corynebacterium ovis on peritoneal macrophages. Infect Immun 12:1439–1449
Hayhurst EJ, Kailas L, Hobbs JK, Foster SJ (2008) Cell wall peptidoglycan architecture in Bacillus subtilis. Proc Natl Acad Sci USA 23:14603–14608
Henderson B, Martin A (2011) Bacterial virulence in the moonlight: multitasking bacterial moonlighting proteins are virulence determinants in infectious disease. Infect Immun 79:3476–3491
Hodgson AL, Bird P, Nisbet IT (1990) Cloning, nucleotide sequence, and expression in Escherichia coli of the phospholipase D gene from Corynebacterium pseudotuberculosis. J Bacteriol 172:1256–1261
Kierek K, Watnick PI (2003) The Vibrio cholerae O139 O-antigen polysaccharide is essential for Ca2-dependent biofilm development in sea water. Proc Natl Acad Sci USA 100:14357–14362
Le Maréchal C, Seyffert N, Jardin J et al (2011) Molecular basis of virulence in Staphylococcus aureus mastitis. PLoS ONE 6e:27354
Modun B, Williams P (1999) The staphylococcal transferrin-binding protein is a cell wall glyceraldehyde-3-phosphate dehydrogenase. Infect Immun 67:1086–1092
Moura-Costa LF, Paule BJA, Freire SM et al (2002) Meio sintético quimicamente definido para o cultivo de Corynebacterium pseudotuberculosis. Rev Bras Saúde Prod An 3:1–9
Norris V, Chen M, Goldberg M, Voskuil J, McGurk G, Holland IB (1991) Calcium in bacteria: a solution to which problem? Mol Microbiol 5:775–778
Ohlsen K, Donat S (2010) The impact of serine/threonine phosphorylation in Staphylococcus aureus. Int J Med Microbiol 300:137–141
Pacheco LG, Slade SE, Seyffert N et al (2011) A combined approach for comparative exoproteome analysis of Corynebacterium pseudotuberculosis. BMC Microbiol 11:12
Pancholi V, Fischetti VA (1992) A major surface protein on group A streptococci is a glyceraldehyde-3-phosphate-dehydrogenase with multiple binding activity. J Exp Med 176:415–426
Paule BJ, Meyer R, Moura-Costa LF et al (2004) Three-phase partitioning as an efficient method for extraction/concentration of immunoreactive excreted-secreted proteins of Corynebacterium pseudotuberculosis. Protein Expr Purif 34:311–316
Rosen R, Sacher A, Shechter N, Becher D, Büttner K, Biran D, Hecker M, Ron EZ (2004) Two-dimensional reference map of Agrobacterium tumefaciens proteins. Proteomics 4:1061–1073
Sarkisova S, Patrauchan MA, Berglund D, Nivens DE, Franklin MJ (2005) Calcium-induced virulence factors associated with the extracellular matrix of mucoid Pseudomonas aeruginosa biofilms. J Bacteriol 187:4327–4337
Sengupta N, Alam SI, Kumar B, Kumar RB, Gautam V, Kumar S, Singh L (2010) Comparative proteomic analysis of extracellular proteins of Clostridium perfringens type A and type C strains. Infect Immun 78:3957–3968
Shimakata T, Minatogawa Y (2000) Essential role of trehalose in the synthesis and subsequent metabolism of corynomycolic acid in Corynebacterium matruchotii. Arch Biochem Biophys 380:331–338
Trost M, Wehmhoner D, Kärs U, Dieterich G, Wehland J, Jansch L (2005) Comparative proteome analysis of secretory proteins from pathogenic and nonpathogenic Listeria species. Proteomics 5:1544–1557
Uchiyama S, Carlin AF, Khosravi A, Weiman S, Banerjee A, Quach D, Hightower G, Mitchell TJ, Doran KS, Nizet V (2009) The surface-anchored NanA protein promotes pneumococcal brain endothelial cell invasion. J Exp Med 31:1845–1852
Vollmer W, Bernard J, Charlier P, Foster S (2008) Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev 32:259–286
Wilson MJ, Brandon MR, Walker J (1995) Molecular and biochemical characterization of a protective 40-kilodalton antigen from Corynebacterium pseudotuberculosis. Infect Immun 63:206–211
Winder FG, Collins PB (1970) Inhibition by isoniazid of synthesis of mycolic acids in Mycobacterium tuberculosis. J Gen Microbiol 66:41–48
Acknowledgments
We are thankful to the Center for Study of Structure and Function of Biomolecules of the Institute of Biological Sciences of the Federal University of Minas Gerais and to the Genomic and Proteomic network of the State of Pará. The current work was supported by CNPq, FAPEMIG and FAPESPA.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Silva, W.M., Seyffert, N., Ciprandi, A. et al. Differential Exoproteome Analysis of Two Corynebacterium pseudotuberculosis Biovar Ovis Strains Isolated from Goat (1002) and Sheep (C231). Curr Microbiol 67, 460–465 (2013). https://doi.org/10.1007/s00284-013-0388-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-013-0388-4