
Abstract
The foodborne pathogen Listeria monocytogenes has the ability to develop biofilm in food-processing environment, which becomes a major concern for the food safety. The biofilm formation is strongly influenced by the availability of nutrients and environmental conditions, and particularly enhanced in poor minimal essential medium (MEM) containing glucose rather than in rich brain heart infusion (BHI) broth. To gain better insight into the conserved protein expression profile in these biofilms, the proteomes from biofilm- and planktonic-grown cells from MEM with 50 mM glucose or BHI were compared using two-dimensional polyacrylamide gel electrophoresis followed by MALDI-TOF/TOF analysis. 47 proteins were successfully identified to be either up (19 proteins) or down (28 proteins) regulated in the biofilm states. Most (30 proteins) of them were assigned to the metabolism functional category in cluster of orthologous groups of proteins. Among them, up-regulated proteins were mainly associated with the pentose phosphate pathway and glycolysis, whereas a key enzyme CitC involved in tricarboxylic acid cycle was down-regulated in biofilms compared to the planktonic states. These data implicate the importance of carbon catabolite control for L. monocytogenes biofilm formation in response to nutrient availability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Biofilms are densely packed multicellular communities of microorganisms attached to a biotic or abiotic surface. It is well known that bacterial cells within biofilms are highly resistant to antibiotics, UV light, acid exposure, dehydration, and phagocytosis in comparison to their free-living (planktonic) forms [12] and are metabolically different from their free-living forms. Listeria monocytogenes, a Gram-positive food borne pathogen responsible for listeriosis, has the ability to form biofilms on food-processing surfaces, which is a major concern for the food safety because bacteria from biofilms can contaminate food products during processing and packaging [7]. Earlier studies and our present data showed that this bacterium preferably produced biofilm in nutrient-limited essential medium rather than the nutrient-rich medium, although the growth rates of cells were dominantly higher in the rich medium [19, 20]. However, the molecular mechanism of nutrient availability as the specific environmental signal to promote biofilm formation is not clear.
To understand the underlying mechanism of biofilm formation of L. monocytogenes adapting to the different nutrient conditions, the differentially expressed proteins obtained from both brain heart infusion (BHI) broth and poor minimal essential medium (MEM) containing 50 mM glucose were investigated in this study using two-dimensional polyacrylamide gel electrophoresis (2-DE) followed by MALDI-TOF/TOF analysis. Our results indicated that carbon catabolite control might be a critical regulatory determinant of bacterial biofilm formation under a variety of cultivation conditions.
Materials and Methods
Bacterial Cultures and Growth of Biofilms
Listeria monocytogenes EGDe (serotype 1/2a) is a gift from Prof. W. Goebel of Department of Microbiology in Wuerzburg University in Germany. For all experiments, individual colonies of the stock culture were inoculated into 5 mL of BHI broth (Difco, Sparks, MD) and incubated at 37 °C with shaking (180 rpm) for overnight, then transferred either directly into the fresh BHI broth with 1:50 dilution, or after three times of washing with the MEM, the pellets were resuspended with 250 mL of MEM [13] and grown at 37 °C with shaking until the optical density at 600 nm reached 0.2, followed by dispensing into microtiter plates or sterile plastic petri dishes.
Microtiter Plate Assay and Fluorescence Microscopic Observation
The microtiter plate assay described previously by Zhou et al. [23] was used to quantify biofilm production of L. monocytogenes EGDe cultured in BHI or MEM at 37 °C after 24, 48, and 72 h.
For fluorescence microscopic observation, biofilm was grown on the glass cover slips which were submerged in a six-well plate (NestBiotech Co., Ltd, Wuxi, China) containing 6 mL of bacterial cultures prepared as described above. The plate was incubated statically at 37 °C for 3, 24, 48 h or 5 days. After the indicated incubation period, slips were taken out carefully and rinsed thrice with phosphate-buffered saline (PBS; pH 7.2) to remove loosely adhered bacteria, and then immersed into 0.1 % fluorescein isothiocyanate (FITC; Sigma-Aldrich) in the dark for 30 min at room temperature to stain the sessile cells in the biofilms. Following staining, slips were rinsed thrice with PBS. Biofilms formed were examined and photographed using Nikon fluorescence microscope E400 (Nikon company, Japan).
Protein Extraction, Two-Dimensional Polyacrylamide Gel Electrophoresis (2-DE)
Biofilms for proteomic analysis were grown on 20 sterile plastic petri dishes (10 cm in diameter) filled with 20 mL of BHI or MEM statically at 37 °C for 48 h. For collection of biofilm-grown cells, the media were discarded and then the dishes were carefully washed thrice with Tris buffer (pH 7.2) to remove loosely adhered bacteria. The cells attached to the dishes were scraped off using a sterile razor and put into 10 mL of Tris buffer and then pelleted by centrifugation (4,000 rpm, 15 min). The planktonic-grown cells (PC) from BHI or MEM medium were grown at 37 °C with shaking (180 rpm) until the optical density at 600 nm reached 0.5. After centrifugation, the pellets were washed thrice with Tris buffer to remove thoroughly the chemical components. For protein extraction, the cell pellets were resuspended in 1.2 mL of pre-cooled sonication buffer containing 7 mol/L urea (Biosharp), 2 mol/L thiourea (Biosharp), 2 % CHAPS (Sigma), 65 mmol/L DTT (Sigma), 0.5 % pharmalyte 3–10 (Bio-Rad), 0.5 mg/mL DNase I (Bio-Rad), 70 U RNase A (Sigma) and 100 mmol/L PMSF (Bio-Rad), 5 μL of protease inhibitor cocktail (Bio-Rad) and 3 g of sterilized silica sands (Sinopharm Chemical Reagent Co. Ltd, China). Cells were sonicated with a UP200S sonifier (Hielscher, Germany) on ice bath eight times for 40 s (20 s pause in between) at power level 5 and 30 % of the active cycle. The cell pellets and silica sands were eliminated by centrifugation at 4 °C for 60 min (13,000×g). The supernatant was collected and stored at −80 °C in aliquots until use. Protein concentrations were determined using Bradford protein assay according to the manufacturer’s instructions (Bio-Rad).
Protein extracts (690 μg) were cup-loaded onto 17-cm strips with a pH range of 4 to 7 (Bio-Rad) for isoelectric focusing, which was performed using a flat-bed Multiphor II (Amersham Biosciences), and then separated on a 12 % SDS-polyacrylamide gel using a Protean II Xi cell (Bio-Rad) as previously reported by Dumas et al. [5]. The gels were stained with Coomassie Brilliant Blue G-250 (Bio-Rad) and scanned on a PowerLook 2100XL scanner (UMAX Technologies).
Image Analysis of the 2-DE Gels and Protein Identification
Image analysis of the 2-DE gels was performed using the PD Quest 7.3.0 software (Bio-Rad) and gel comparisons were performed through a master gel that was a synthetic image containing all the protein spots of all the gels to be analyzed. Spot intensities were normalized to the sum of intensities of all valid spots in the master gel. Each protein spot was thus identified by a unique master number. Only reliable protein spots were retained for analysis. A protein spot was considered reliable when it was present or absent at least in two gels out of three repetitive gels for a strain. For analysis of changes in protein expression in the biofilm or planktonic states under BHI or MEM media, a protein was considered differentially produced when changes in normalized spot intensities were of least twofold at a significance level of P < 0.05 (Student’s t test for paired samples). Three independent biological replicates were carried out under the same condition.
Spots of interest were subjected to tryptic in-gel digestion and analyzed by a MALDI-TOF/TOF mass spectrometer (ABI 4700 Proteomics Analyzer; Applied Biosystems). For data acquisition and export, two software packages, 4000 Series software and GPS Explorer™ v3.5, provided by the ABI were used. After data acquisition, files were uploaded for searching the NCBI (nr) L. monocytogenes EGD-e database with the following selection parameters: trypsin digestion allowing up to one missed cleavage, methionine oxidation, cysteine carbamidomethyl and peptide tolerance of ±0.1 Da. Only proteins of their protein score CI % ≥95% were considered to be correctly identified.
Results
Formation of Biofilm was Enhanced in Glucose Minimal Essential Medium
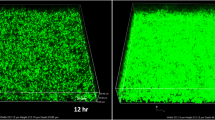
To investigate whether the culture media has an impact on biofilm formation, L. monocytogenes EGDe was cultured in both in rich BHI and poor glucose MEM media. As shown in Fig. 1b and C, L. monocytogenes EGDe was able to develop biofilm with meshwork of bacterial aggregates in those media. Dense localized biofilm structures formed within 24 h and became denser after 24 h. Thicker and complex biofilm structures were formed after 48 h of incubation (Fig. 1c). However, biofilm biomass generated in MEM was much greater than that in a parallel BHI culture inoculated at the same period, while the growth rates of cells in BHI were actually higher than those in MEM (Fig. 1a, b). The fluorescence microscopy also showed that the biofilm formed in MEM had more clumped and aggregated cells than that in BHI even in the early phase of biofilm development (3 h), which were well consistent with the quantitative estimations of biofilms.

Biofilm formation by L. monocytogenes EGDe in BHI and MEM with 50 mM glucose. a Cell densities after 24, 48, and 72 h incubation. b Biofilm productions measured by microtiter plate assay after 24, 48, and 72 h incubation. c Fluorescence microscopy observation of L. monocytogenes EGDe biofilm formation. The strain was grown at 37 °C in BHI (I–IV) or MEM with 50 mM glucose (V–VIII) on glass slips for 3, 24, 48 h, and 5 days. The error bars represent the standard deviations of triplicate experiments (when not visible, error bars are smaller than the thickness of the bar border). Bars equal 0.1 mm
Identification of Proteins Differentially Expressed Under Biofilm Formation Conditions
Given that L. monocytogenes can develop biofilm in different nutrient conditions, we therefore propose that a conserved signaling pathway for nutrient metabolism exists in L. monocytogenes biofilm formation under different environments, in which the capability of biofilm development in different nutrient conditions would be due to differential regulation of this conserved signal pathway. To test this hypothesis, the differentially expressed proteins common in two biofilm states cultivated under rich BHI broth and poor MEM supplemented with 50 mM glucose were investigated in the present study using 2-dimensional polyacrylamide gel electrophoresis. Figure 2a–d shows representatives of 2-DE patterns for L. monocytogenes wild-type strain EGDe grown in MEM or BHI under biofilm and planktonic states. A total of 47 proteins were successfully identified to be differentially expressed in both biofilm states (BHI and MEM) compared to their corresponding planktonic states, 19 of which were up-regulated or only detected in the biofilm states, and 28 were down-regulated or not found in the protein profiles from biofilm-grown cells (Fig. 1; Table 1). Meanwhile, five proteins identified were found in more than one spot, indicating that the protein isoforms or posttranslational modification events occurred.

2-DE gels of cytoplasmatic extracts of L. monocytogenes EGDe from biofilm-grown cells in MEM with 50 mM glucose (a) or BHI (b), and planktonic-grown cells cultured in MEM with 50 mM glucose (c) or BHI (d). Spots identified by mass spectrometry are labeled and their identification is listed in Table 1
Based on analysis of the cluster of orthologous groups of proteins (COG) obtained from the MicrobesOnline Comparative Genomics Database (MOCGD) (http://www.microbesonline.org/), these proteins can be classified into 11 functional categories (Table 1), and most of them (30 proteins) were involved in energy production (7 proteins), metabolism of amino acid (7 proteins), nucleotide (6 proteins), carbohydrate (9 proteins), and lipid (1 protein). Remarkably, several key enzymes involved in the pentose phosphate pathway (6-phosphogluconate dehydrogenase encoded by lmo1376, 6-phosphogluconolactonase encoded by pgl and transketolase encoded by tkt) and the 6-phosphofructokinase encoded by pfkA in glycolysis were present at a higher level in biofilms, while the isocitrate dehydrogenase encoded by citC, which converts isocitrate to alpha ketoglutarate in the tricarboxylic acid (TCA) cycle, was down-regulated in biofilm-growth cells compared with planktonic-growth cells. These data suggest that TCA activity may be present to a low level in L. monocytogenes biofilms, so that energy will be supplied from glycolysis and the pentose phosphate pathways (see “Discussion”). Interestingly, expression of two enzymes, Sod (superoxide dismutase encoded by lmo1439) and MsrA (methionine sulfoxide reductase A encoded by lmo1860), which play a role in cellular defense against oxidative stress, was also repressed in biofilms. Since an increased ratio of TCA cycle to respiratory activity boosts the production of reactive oxygen species (ROS) such as O2− and subsequently H2O2, i.e., oxidative stress, low expression level of Sod and MrsA may be due to relatively low TCA activity. In fact, Nguyen et al. [11] reported that the activity of Sod was reduced in response to the repressed respiration in oral Streptococci biofilm and considered the low respiratory activity would facilitate bacterial cells to survive in the crowded biofilm condition. Moreover, enzymes related to degradation of protein, peptides, glycopeptides (Lmo2188, Lmo1611, and Lmo1138), and mRNA (PnpA) were repressed in biofilms, while those involved in protein synthesis were up-regulated, particularly, in cell wall biogenesis, including elongation factors G and Ts, 50S ribosomal protein L10, Lmo0197, and Lmo2391. Furthermore, a glutamine synthetase encoded by glnA was also up-regulated. Glutamine synthetase necessary for cell wall resistance and biofilm formation of Mycobacterium bovis [2] has been found to be synthesized in higher amounts in the proteomic study of Staphylococcus aureus biofilm cells [14]. In addition, two proteins GuaB and Upp were detected to be up-expressed in L. monocytogenes biofilm in this study. GuaB that converts IMP into GMP was important for the production of Mycobacterium avium biofilm [22], and Upp that catalyzes uracil to UMP was required to biofilm development in Pseudomonas aeruginosa through control of quorum-sensing (QS) pathways [21]. Upp was aslo found to be induced by glucose starvation in L. monocytogenes biofilm [8]. These differentially expressed proteins have also been identified as differentially expressed during biofilm formation by other bacteria, suggesting that there may be widely conserved responses of bacterial cells to biofilm formation.
Discussion
In this study, proteomics was applied to gain insight into key cellular events that allow L. monocytogenes to develop mature biofilms under nutrient-rich (BHI) and poor (MEM) conditions. We observed that the biofilm-forming processes mainly affected metabolic pathways for energy production and transport as well as metabolism of amino acid, nucleotide, and carbohydrate. Interestingly, many proteins related to the pentose phosphate pathway and glycolysis were found to present in a higher level, while a key enzyme CitC involved in TCA cycle were down-regulated in biofilms compared to the planktonic states. The enzymatic activity of the TCA cycle has been found to be negatively associated with the synthesis of the biofilm-promoting exopolysaccharide polysaccharide intercellular adhesin (PIA) in S. aureus and S. epidermidis, in which the activated TCA cycle decreased PIA synthesis, reduced biofilm formation, and significantly attenuated virulence in an in vivo biofilm model [16, 24]. This negative regulation of entrance to the TCA cycle was considered to allow the biofilm-growth cells to avoid the production of excess ATP as long as they can obtain enough ATP through glycolysis and the pentose phosphate pathways (i.e., substrate level phosphorylation) [16, 24]. In addition, while some specific determinants including flagella proteins [9] and the Agr system for the putative QS peptide [15] for L. monocytogenes biofilm formation were not detected in this study, this might be due to the detection limit of the proteomic technique and nonoptimal temperature (37 °C) for the expression of listerial flagella proteins used in this study. However, the transcription and/or stability of RNAIII, part of the Agr QS system, was increased in an S. aureus TCA cycle mutant [24], raising the possibility that decreased TCA cycle activity could increase bacteria biofilm development via alteration of RNAIII transcription levels.
A significant number of reports have demonstrated that a specific environmental signal nutrient availability plays a critical role in promotion of L. monocytogenes biofilm formation [3, 4]. Specifically, bacterial biofilm mass is enhanced during growth in minimal essential media containing glucose [3, 19], suggesting the global catabolite regulation (CR) may influence bacterial biofilm formation. Similar to other low G + C content gram-positive bacteria, such as Bacillus subtilis and S. aureus, in L. monocytogenes, the predominant mechanism of CR involves transcriptional repression mediated by the catabolite control protein A (CcpA) [1]. After formation of a complex with the phosphorylated co-repressors, HPr, one of the components of the phosphoenolpyruvate:sugar phosphotransferase (PTS) sugar uptake system, CcpA becomes active and blocks transcription initiation via binding to the catabolite responsive elements (cre) located in or near carbon catabolite repressed promoters. The phosphorylation of Hpr is controlled by the activity of a kinase that responds to the intracellular concentration of fructose 1-6-bisphosphate (FBP), which varies based on the rate of glucose transport into the cell [1]. High concentration of FBP can activate the ATP-dependent HPr kinase to phosphorylate HPr and form a complex with CcpA. CcpA has been shown to coordinate central metabolism and biofilm formation in some gram-positive bacteria, such as B. subtilis, S. aureus, and S. epidermidis. In B. subtilis, CcpA enhances pyruvate assimilation and shut-down of the TCA cycle and respiration [6]. In S. aureus and S. epidermidis, CcpA functions as a regulator of the TCA cycle, deletion of ccpA derepressed TCA cycle activity and inhibited biofilm formation [16, 17]. In this study, two important proteins closely related to up-regulation of CcpA activity were detected: the 6-phosphofructokinase encoded by pfkA which catalyzes the formation FBP from d-fructose 6-phosphate, was up-regulated; and Lmo0783 (MpoB), a putative mannose-specific PTS component IIB, which is one of components of the major glucose uptake systems in L. monocytogenes [18] and negatively controlled by CcpA [10], was down-regulated. These results indicated that the activity of CcpA in the mature biofilms might be higher than that in the planktonic state. Hence, the observation that formation of biofilm by L. monocytogenes was enhanced during growth in glucose MEM appear partially due to the relatively higher level of CcpA-mediated CR in MEM than that in BHI, since the growth of L. monocytogenes in nutrient-rich media depends only in part on CcpA-regulated carbohydrate metabolism [18]. Therefore, although the impact of up-expression of the pyruvate dehydrogenase complex (except PdhC; this complex was encoded by pdhABCD operon that oxidates pyruvate to acetyl-CoA) in biofilms remains to be unclear and needs to be further investigated, given the importance of coordination of carbon source utilization and regulation of metabolic pathways, it is reasonable to assume that CcpA could have a positive role in regulation of genes that are important for biofilm formation by L. monocytogenes. This hypothesis needs to be further investigated, while the absence of CcpA protein identified in this study can be due to the detection limit of the proteomic technique. We are currently working on this to confirm our proposal.
References
Behari J, Youngman P (1998) A homolog of CcpA mediates catabolite control in Listeria monocytogenes but not carbon source regulation of virulence genes. J Bacteriol 180:6316–6324
Chandra H, Basir S, Gupta M, Banerjee N (2010) Glutamine synthetase encoded by glnA-1 is necessary for cell wall resistance and pathogenicity of Mycobacterium bovis. Microbiology 156:3669–3677
Chang Y, Gu W, Fischer N, McLandsborough L (2012) Identification of genes involved in Listeria monocytogenes biofilm formation by mariner-based transposon mutagenesis. Appl Microbiol Biotechnol 93(5):2051–2062
Djordjevic D, Wiedmann M, McLandsborough L (2002) Microtiter plate assay for assessment of Listeria monocytogenes biofilm formation. Appl Environ Microbiol 68:2950–2958
Dumas E, Desvaux M, Chambon C, Hébraud M (2009) Insight into the core and variant exoproteomes of Listeria monocytogenes species by comparative subproteomic analysis. Proteomics 9:3136–3155
Fujita Y (2009) Carbon catabolite control of the metabolic network in Bacillus subtilis. Biosci Biotechnol Biochem 73:245–259
Gandhi M, Chikindas M (2007) Listeria: a foodborne pathogen that knows how to survive. Int J Food Microbiol 113:1–15
Helloin E, Jansch L, Phan-Thanh L (2003) Carbon starvation survival of Listeria monocytogenes in planktonic state and in biofilm: a proteomic study. Proteomics 3:2052–2064
Lemon K, Higgins D, Kolter R (2007) Flagellar motility is critical for Listeria monocytogenes biofilm formation. J Bacteriol 189:4418–4424
Mertins S, Joseph B, Goetz M, Ecke R, Seidel G, Sprehe M, Hillen W, Goebel W, Müller-Altrock S (2007) Interference of components of the phosphoenolpyruvate phosphotransferase system with the central virulence gene regulator PrfA of Listeria monocytogenes. J Bacteriol 189:473–490
Nguyen P, Abranches J, Phan T, Marquis R (2002) Repressed respiration of oral streptococci grown in biofilms. Curr Microbiol 44:262–266
O’Toole G, Kaplan H, Kolter R (2000) Biofilm formation as microbial development. Annu Rev Microbiol 54:49–79
Premaratne R, Lin W, Johnson E (1991) Development of an improved chemically defined minimal medium for Listeria monocytogenes. Appl Environ Microbiol 57:3046–3048
Resch A, Leicht S, Saric M, Pásztor L, Jakob A, Götz F, Nordheim A (2006) Comparative proteome analysis of Staphylococcus aureus biofilm and planktonic cells and correlation with transcriptome profiling. Proteomics 6:1867–1877
Riedel C, Monk I, Casey P, Waidmann M, Gahan C, Hill C (2009) AgrD-dependent quorum sensing affects biofilm formation, invasion, virulence and global gene expression profiles in Listeria monocytogenes. Mol Microbiol 71:1177–1189
Sadykov M, Hartmann T, Mattes T, Hiatt M, Jann N, Zhu Y, Ledala N, Landmann R, Herrmann M, Rohde H, Bischoff M, Somerville G (2011) CcpA coordinates central metabolism and biofilm formation in Staphylococcus epidermidis. Microbiology 157:3458–3468
Seidl K, Goerke C, Wolz C, Mack D, Berger-Bächi B, Bischoff M (2008) Staphylococcus aureus CcpA affects biofilm formation. Infect Immun 76:2044–2050
Stoll R, Mertins S, Joseph B, Müller-Altrock S, Goebel W (2008) Modulation of PrfA activity in Listeria monocytogenes upon growth in different culture media. Microbiology 154:3856–3876
Todhanakasem T, Young G (2008) Loss of flagellar-based motility by Listeria monocytogenes results in the formation of hyperbiofilms. J Bacteriol 190:6030–6034
Tsai H, Hodgson D (2003) Development of a synthetic minimal medium for Listeria monocytogenes. Appl Environ Microbiol 69:6943–6945
Ueda A, Attila C, Whiteley M, Wood T (2009) Uracil influences quorum sensing and biofilm formation in Pseudomonas aeruginosa and fluorouracil is an antagonist. Microb Biotechnol 2:62–74
Yamazaki Y, Danelishvili L, Wu M, Macnab M, Bermudez L (2006) Mycobacterium avium genes associated with the ability to form a biofilm. Appl Environ Microbiol 72:819–825
Zhou Q, Feng F, Wang L, Feng X, Yin X, Luo Q (2011) Virulence regulator PrfA is essential for biofilm formation in Listeria monocytogenes but not in Listeria innocua. Curr Microbiol 63:186–192
Zhu Y, Xiong Y, Sadykov M, Fey P, Lei M, Lee C, Bayer A, Somerville G (2009) Tricarboxylic acid cycle-dependent attenuation of Staphylococcus aureus in vivo virulence by selective inhibition of amino acid transport. Infect Immun 77:4256–4264
Acknowledgments
This work was funded by the National Natural Science Foundation of China (30970111), Excellent Youth Foundation of Hubei Scientific Committee (No. 2009CDA124) and CCNU Project (No. CCNU09Y01001).
Author information
Authors and Affiliations
Corresponding author
Additional information
Qingchun Zhou and Xiaoqin Feng contributed equally to this work and should be considered as co-first authors.
Rights and permissions
About this article
Cite this article
Zhou, Q., Feng, X., Zhang, Q. et al. Carbon Catabolite Control is Important for Listeria monocytogenes Biofilm Formation in Response to Nutrient Availability. Curr Microbiol 65, 35–43 (2012). https://doi.org/10.1007/s00284-012-0125-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-012-0125-4