
Abstract
The bacteriophage μ1/6 integrates its DNA into the chromosome of tetracycline producing strains of Streptomyces aureofaciens by a site-specific recombination process. A bioinformatic analysis of the μ1/6 genome revealed that orf5 encodes a putative integrase, a basic protein of 416 amino acids. The μ1/6 integrase was found to belong to the integrase family of site-specific tyrosine recombinases. The phage attachment site (attP) was localized downstream of the int gene. The attachment junctions (attL and attR) were determined, allowing identification of the bacterial attachment site (attB). All attachment sites shared a 46-bp common core sequence within which a site-specific recombination occurs. This core sequence comprises the 3′ end of a putative tRNAThr gene (anticodon TGT) which is completely restored in attL after integration of the phage into the host genome. An integration vector containing μ1/6 int-attP region was inserted stably into the S. aureofaciens B96, S. lividans TK24, and S. coelicolor A3. The μ1/6 integrase was shown to be functional in vivo in heterologous Escherichia coli without any other factors encoded by Streptomyces. In vitro recombination assay using purified μ1/6 integrase demonstrated its ability to catalyze integrative recombination in the presence of a crude extract of E. coli cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Integration of the phage genome into the host chromosome is accomplished by a phage integrase enzyme that mediates a site-specific recombination between the phage attachment site (attP) and the bacterial attachment site (attB) [6]. Site-specific recombinases can be classified into two families: the tyrosine recombinase (lambda integrase) family and the serine recombinase (resolvase/invertase) family based on amino acid sequence similarities and catalytic residues, either tyrosine or serine. These enzymes use a tyrosine or serine as the nucleophile that breaks specific phosphodiester bonds and covalently attaches the enzyme to the DNA. The recombination reaction mechanism used by tyrosine integrases involves the formation and resolution of a Holliday junction intermediate, while serine recombinases act via a simultaneous, four-strand staggered break, rotation, and religation of cleaved DNA substrates. Properties such as the specificity and effectivness make phage integrases suitable for the engineering of DNA in genomes of both homologous and heterologous organisms [15, 16, 34].
Streptomyces are Gram positive, sporulating soil bacteria characterized by a complex secondary metabolism producing antibiotic compounds and other metabolits with medical properties. Several integration systems based on phage integrases have been utilized for genetic manipulation of this industrially important microorganisms [14, 18]. Recently, integrases of Streptomyces phages R4 [25], φC31 [32], and φBT1 [8] from the serine recombinase family have been shown to function in mammalian cells. They are now being tested for their effectivness in various human cells as a new tool for gene therapy [7]. First example of medically relevant application have been demonstrated for the φC31integrase, which was used to promote site-specific integration of the dystrophin gene into the patient’s own myogenic precursor cells [27].
Bacteriophage μ1/6 has a narrow host range for industrially important S. aureofaciens strains producing tetracycline. Its ds DNA genome contains 38,194 bp with a G + C content of 71.2%. The analysis of the entire sequence revealed the presence of a putative integrase gene [11]. Here, we describe the genetic elements involved in site-specific integration of the phage μ1/6 into the bacterial chromosome, i.e., the integrase gene, the phage attP, and the bacterial attB attachment sites. An integration vector based on the integration cassette was shown to be functional in S. aureofaciens, S. coelicolor, and S. lividans. Furthermore, the site-specific recombination system based on μ1/6 integrase was found to work also in heterologous Escherichia coli.
Materials and Methods
Bacteria, Phage, and Plasmids
Escherichia coli strains were grown in Luria-Bertani medium (LB). When necessary, the medium was supplemented with appropriate antibiotics: ampicillin (Ap) 100 μg/ml, chloramphenicol (Cm) 25 μg/ml, and kanamycin (Kn) 50 μg/ml. Bacteriophage μ1/6 was propagated on tetracycline producing Streptomyces aureofaciens B96 [10]. Streptomycetes were grown in TSB medium with 0.7% of glycine, or in YEME medium [18]. Preparation of protoplasts of S. lividans TK24 and S. coelicolor A3 cells (both strains are from John Innes Culture Collection, Norwich, UK), and transformation procedure was performed as described previously [18]. Transformants were selected by means of an overlay with 3 ml of soft agar containing thiostrepton (Tsr) at 500 μg/ml. Protoplasts of S. aureofaciens B96 were prepared by a method mentioned above with some modifications. The streptomycete culture was first grown overnight in TSB medium, followed by a 48 h cultivation in YEME medium. The incubation time of the harvested mycelium in lysozyme solution was extended to 2.5–3 h and the concentration of lysozyme was increased to 2 mg/ml. Transformation of E. coli cells was carried out by the RbCl method [29].
DNA Manipulations
Bacteriophage DNA was prepared according to [10]. Plasmid DNA isolation, digestion of DNA with restriction enzymes, ligation, and other DNA manipulation procedures were performed following standard methods [29], or supplier’s instructions. Total DNA of Streptomyces cells was isolated as described previously [18].
Construction of the Integration Vector
For construction of the integration vector pCTint, a MluI/SauI deletion derivative (ΔpCT) of the replicon probe vector pIMB03 [13] was used. This plasmid contains E. coli replicon p15A, Cm resistance gene for selection in E. coli, and thiostrepton resistance gene for selection in streptomycetes. A fragment containing int-attP region (nucleotides 5,332–3,659; GenBank DQ372923) was amplified from μ1/6 DNA using primers INTF (5′CCAAGCTTGCTGCTGTGGGCAGCCTCTGCGGC3′) and INTR (5′GAAGATCTCGCACAAGGCCGCCCCTACAGG3′) deduced from the bioinformatic analysis of the μ1/6 sequence (restriction sites used for cloning are underlined in oligos). After HindIII/BglII digestion, the PCR fragment was subcloned into HindIII/BglII cleaved ΔpCT. Ligation mix was introduced into E. coli MC1061 (The Coli Genetic Stock Centre, Yale University, USA). The resulting plasmid, designated pCTint, was used to transform protoplasts of S. aureofaciens B96, S. coelicolor A3, or S. lividans TK24. Transformants were then selected for resistance to thiostrepton.
Hybridization Experiments
Total DNA isolated from Streptomyces strains with or without the integrated vector was cleaved with the appropriate restriction enzymes and DNA fragments were transferred from agarose gels to nylon membranes Hybond-N+ (Amersham, GE Healthcare, UK). Probes were labeled with digoxigenin-11-dUTP using the Dig DNA Labeling and Detection Kit (Roche Applied Sciences, Germany). Hybridization experiments were performed according supplier’s specification.
Identification of attL, attR, and attB
To identify attL and attR sequences, protoplasts of S. aureofaciens B96 were transformed with integration vector pCTint. The chromosomal DNA of S. aureofaciens B96:pCTint was cut with PstI (no site present in the pCTint sequence) and the DNA fragments were ligated under diluted conditions (5 μg/ml) to favor intramolecular annealing. The ligation mix was used to transform E. coli strain MC1061. The resulting plasmid isolated from Cmr transformants was designated pB96CTint. Flanking chromosomal regions of S. aureofaciens B96 present in pB96CTint contained attL and attR which were determined as follows: PstI/HindIII and PstI/BglII fragments were ligated into PstI/HindIII and PstI/BamHI cleaved pBluescriptSK+ (Stratagene, USA), respectively, and subsequently sequenced. A fragment containing attB was amplified from S. aureofaciens B96 genomic DNA using primers BF (5′CCCAAGCTTATCGGTTCCATCACGCTCAC3′) and BR (5′CGGGATCCCCAAGACCCTGGTCAAGAACG3′) complementary to attL and attR, digested with HindIII and BamHI (sites underlined in oligos). The resulting PCR product (399 bp) was inserted into pBluescript SK+ cleaved with the same restriction enzymes and sequenced.
In Vivo Integration Assay in E. coli
The fragment containing int gene and attP sequence (see above) was digested with HindIII and BglII and inserted into the plasmid LITMUS 38 (New England Biolabs, USA) cleaved with HindIII and BamHI. After transformation of E. coli XL-1 blue (Stratagene, USA) with the DNA, the resulting plasmid LITint-attP was obtained from Apr transformants. Plasmid pACYCattB was constructed by amplifying the attB fragment from μ1/6 DNA using primers (BR and BR), digesting the product with HindIII and BamHI, and inserting the 399-bp fragment digested with the same enzymes into pACYC184 (New England Biolabs, USA). E. coli transformants harboring the resulting plasmid pACYCattB were selected on LB plates with Cm (25 μg/ml). Plasmids LITint-attP and pACYCattB were cotransformed into E. coli XL-1 blue and transformants were selected on LB plates containing Ap (100 μg/ml) and Cm (25 μg/ml). Apr Cmr colonies were grown overnight in liquid LB medium containing the above mentioned antibiotics. Plasmids were isolated by alkaline lysis [4]. The recombination event was confirmed by restriction analysis of plasmids LITint-attP, pACYCattB and the recombinant product with HpaI, XbaI, and HpaI/XbaI, respectively. The smaller XbaI/HpaI fragment was recovered from agarose gel, cloned into pBluescript SK+ digested with XbaI and EcoRV, and sequenced. The second junction site was confirmed by PCR amplification using recombinant product with following primers: RF (5′ACGACCGCGGGTCCTCGGTCAC3′) and RR (5′CCAAGACCCTGGTCAAGAACG3′). The resulting PCR product was sequenced.
In Vitro Integration Assay
In vitro recombination assay was performed as described previously [22] with some modifications. Plasmid pBSKattP was constructed by amplifying the attP region from μ1/6 DNA using primers AF (5′CCCAAGCTTGGTCCTCGGTCACGTGGCTGTACCA) and AR (5`CGGAATTCCGCACAAGGCCGCCCCTACAGG). The 470-bp PCR fragment was digested with HindIII/EcoRI (sites underlined in oligos) and inserted into pBluescript SK+ cleaved with the same enzymes. The intermolecular integration reaction utilized approximately 0.03 pmol of supercoiled plasmid pBSKattP (3.4 kbp) isolated by the CsCl centrifugation method [29] and 0.3 pmol of a linear attB fragment (399 bp). Reactions also contained 20 mmol/l Tris–Cl pH 7.5, 25 mmol/l NaCl, 1 mmol/l DTT, 10 mmol/l spermidine, 1 mg/ml BSA, 2–4 μl of bacterial cell extract, and 2 μl (approximately 1 μg) of purified integrase protein in a total volume of 20 μl. Recombination reactions were incubated overnight at 30°C, heat killed at 75°C for 10 min, and electrophoresed through a 0.8% agarose gel.
The μ1/6 integrase was overexpressed in E. coli BL21(DE3) using the pET system. The enzyme was purified to near homogeneity by metal-affinity chromatography for His-tagged proteins (Supplementary Material).
The bacterial cell extract was prepared as follows: The culture of E. coli MC1061 cells was grown in 50 ml LB medium until it reached the late logarithmic phase. Cells were harvested by centrifugation and resuspended in 5 ml of buffer containing 20 mmol/l Tris–Cl pH 7.8, 5 mmol/l EDTA, 100 mmol/l NaCl, 1 mmol/l DTT. After sonication, the cell extract was clarified by centrifugation at 10,000×g and used in recombination reaction.
Results and Discussion
Integrase Gene on the μ1/6 Genome
Bioinformatic analysis of the μ1/6 sequence [11] revealed at least 52 putative protein coding regions. The μ1/6 genome is apparently organized into two oppositely transcribed arms. The left shorter arm transcribed leftwards on the genetic map encompasses the genes 2–7 involved probably in the life cycle regulation and phage integration (lysogeny). A homology search indicated that the orf5 encodes a putative integrase of phage μ1/6. The predicted integrase is a basic protein of 416 amino acids, whose estimated molecular mass and pI are 47.6 kDa and 9.6, respectively. As for λ phage, the orf 5 is located in a cluster of genes transcribed leftwards on the genome, i.e., in the opposite direction to the majority of the phage genes. The gp5 displays a very significant homology to the integrase gp61 of S. venezuelae phage VWB [33]. Comparison of gp5 protein sequence with known integrases revealed that it belongs to the diverse tyrosine recombinase/integrase family, including λ integrase, bacteriophage P1 Cre recombinase, and bacterial XerD/C recombinase. The N-terminal domain of tyrosine recombinases includes residues responsible for binding to DNA located away from the recombination points. These arms sites contribute importantly to the direction and specificity of integrases, and consequently differ from system to system [5, 9]. The C-terminal domain binds the lower affinity core-type sites and contains the catalytic site. Members of this family were previously found to share four strongly conserved residues located in the C-terminal part of the protein sequences [1, 2]. These residues consist of arginine-histidine-X-X-arginine, and a catalytic tyrosine located closest to the C-terminus. Besides the conserved tetrad R-H-R-Y, located in boxes I and II, several newly identified specific sequence patches, including charged amino acids, contribute to the overall protein fold [24]. The sequence alignment of C-terminal part of the μ1/6 integrase against those of several integrase proteins identified the structurally important and conserved regions (Fig. 1). The Box I contains the Arg residue (R223), whereas Box II includes the His-X-X-Arg motif (H353, R356) and the Tyr residue (Y389), which has been presumed to form covalently an intermediate between recombinase and the DNA.

Alignment of the carboxyl terminal region of μ1/6 integrase (gp5) with homologous proteins: Streptomyces phage VWB gp61 (GenBank AA229749), Micromonospora carbonacea phage pMLP1 IntM (GenBank AA046045), Mycobacteriophage Ms6 Int (GenBank AAD03774), S. avermitilis Int8 (GenBank BAC71440), tyrosine recombinase XerD from E. coli (GenBank AAA62787), and λ phage Int (GenBank AAA96562). The four conserved residues required for the catalytic activity [1, 2] are indicated below the sequences. The structurally important and conserved regions, Box I and Box II [2], and Patches I–III [24], are represented by a line above the sequences. Identical or highly similar amino acid residues are indicated as white letters shadowed with black, and other functionally similar residues are highlighted in gray. The secondary structure of the C-terminal domain of λ integrase [21] is shown under the alignment; the letters (h) and (s) represent α-helix and β-sheet structures, respectively. The alignment was performed using ClustalW program (http://www.ebi.ac.uk/clustalw/)
Determination of attP, attL, attR, and attB Region Sequences
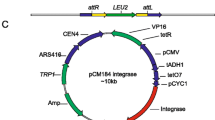
Commonly, the functions required for integration (i.e., attP and integrase) are tightly clustered, thus the phage attP site was deduced from the analysis of the sequences adjacent to the previously identified integrase gene of the μ1/6 phage. To assay integration activity of μ1/6 Int, the integration vector pCTint was constructed (Fig. 2a). The thiostrepton resistance gene on this non-replicative plasmid was used as a selection marker in streptomycete to test whether int-attP region was sufficient to mediate site-specific recombination in vivo. After transformation of protoplasts of S. aureofaciens B96 cells, Tsrr transformants were obtained at a frequency of about 30–50 per μg DNA. Total DNA of six randomly chosen Tsrr transformants was digested with appropriate restriction endonucleases and was analyzed by Southern blot hybridization using the plasmid pCTint as a probe. Hybridization data indicated that integration of the plasmid pCTint was site-specific (Fig. 2b). The results proved that the int-attP region of phage μ1/6 in the integration vector pCTint contained all information required for the recombination process. The μ1/6 int-attP-containing vector was also efficiently integrated into the chromosome of S. lividans and S. coelicolor. Stable Tsrr integrants were recovered and confirmed by Southern hybridization (data not shown).

a A physical map of the integration vector pCTint. Localization of the gene encoding the μ1/6 integrase (int), attP loci, and origin of replication in E. coli (ori p15A) are shown. The Cm resistance and the Tsr resistance were used as selection marker in E. coli and Streptomyces, respectively. b Integration of plasmid pCTint into S. aureofaciens B96 analyzed by Southern hybridization experiments. Chromosomal DNA of S. aureofaciens B96:pCTint was digested with (1) KpnI (one site in pCTint), (2) BamHI (no site in pCTint), (3) total DNA of S. aureofaciens B96 digested with BamHI, (M) labeled DNA size marker (λ + StyI). Plasmid pCTint was used as a probe
Analysis of the predicted attP site showed that the latter was located downstream of the int gene in a non-coding region. Within attP, a complex array of repeats was observed (Fig. 3a). Two direct repeats of 9 bp (DR1) and 8 bp (DR2) were situated upstream of the core sequence. Two another direct repeats DR3 (11 bp with one mismatch) and DR4 (6 bp) were observed downstream of the core. An inverted repeat sequence (IR) of 14 bp with an interval of an odd number of bp was also found downstream of the common core. This 14 bp IR could represent a weak transcriptional terminator of the int gene. These repeats are likely to represent binding sites for the integrase (arm-type site) and other factors involved in recombination.

a Nucleotide sequence downstream of the μ1/6 integrase gene (int) containing the attP region. The C-terminal amino acid sequence from integrase is given below the nt sequence. The core sequence is underlined. Arrows indicate direct (DR) and inverted (IR) repeats. b Nucleotide sequence of the S. aureofaciens B96 attB region. The core sequence is underlined. The potential tRNAThr gene is boxed. The anticodon TGT is shaded in black. Arrows indicate inverted repeats. c Alignment of recombination attachment sites, attB (bacterial attachment site), attL (left phage-host junction site), attR (right phage-host junction site), and attP (phage attachment site). The 46-bp common core sequence is underlined. The tRNAThr genes found both in attB and attL are boxed
It was found that integration of the phage μ1/6 occured within a 1.5 kbp chromosomal PstI fragment of S. aureofaciens B96 (data not shown). The integration vector pCTint was used to determine the attL and attR region sequences which are generated after phage or plasmid integration into the bacterial chromosome. The fragment containing both the integration vector and the flanking chromosomal regions was obtained by PstI digestion of total DNA from S. aureofaciens B96:pCTint and ligation to obtain circular molecules. Transformation of E. coli MC1061 yielded four Cmr clones, all containing ≈7 kb plasmid named pB96CTint. According to the restriction analysis of pB96CTint, the flanking chromosomal regions were cloned and sequenced. On the basis of the attL and attR nucleotide sequences, two new primers were designed and used to amplify the attB region of non-lysogenic S. aureofaciens B96 (Fig. 3b). The sequence comparison (Fig. 3c) revealed a 46-bp region common to all attachment sites. It represented the core where the strand exchange occurs during the prophage formation. The DNA sequence of the attB region was investigated for similarities with databases entries. The sequence showed strong similarities with the tRNAThr gene (anticodon TGT) of Arthrobacter (95% homology), S. avermitilis (92%), S. coelicolor (92%), S. scabiei (92%), S. bingchenggensis (87%). A tRNA search revealed the presence of tRNA cloverleaf secondary structure in attB and attL. The 46-bp identical core sequence comprises the 3′ terminal part of tRNAThr gene in the bacterial chromosome, thus permitting the recovering of an intact copy of this gene in the lysogenized strain. Like other tRNA genes within the genus Streptomyces [30], the tRNAThr that serves as the attachment site for phage μ1/6 appears to be a class II-type tRNA gene requiring post-transcriptional modification by nucleotidyltransferase to acquire 3′ CCA end. The inverted repeat sequence downstream of the tRNA gene has been found, for example, in cases of streptomycete plasmid integration systems [17], S. rimosus phage RP3 [12], or S. venezualae phage VWB [33]. This region of dyad symmetry might function as a terminator that prevents cotranscription of phage genes with the tRNA gene after the phage integration.
Integration into Heterologous E. coli Cells
To test whether the μ1/6 integration module was functional in Gram-negative bacteria, we used a similar assay system that was previously developed for serine integrase φMR11 [28]. Integration reaction shown in Fig. 4a was established using plasmid LITint-attP composed of int-attP region and the compatible plasmid pACYCattB carrying the attB site. Both plasmids were cotransformed into E. coli XL1-blue cells. Restriction analysis od plasmids isolated from clones resistent to both antibiotics (Ap, Cm) revealed the presence of recombinant product, designated pRec (Fig. 4b). The recombination event was further confirmed by sequencing of the smaller fragment obtained after digestion of the recombinant product pRec. Sequencing data showed the presence of attL in this fragment (Fig. 4c). The second junction site, attR, on pRec was confirmed by PCR amplification and sequencing of the PCR product (not shown). These results exactly corresponded to the integration reaction scheme shown in Fig. 4a. Previously described phage integrases of the tyrosine family require a factor encoded by their host for recombination [16]. In the λ system, integration host factor (IHF) functions by binding to sites within the attP and bending the DNA. In this way, the two domains of Int are simultaneously bound to core- and arm-type sites [19, 20]. Integrase μ1/6, clearly a member of the λ integrase family, was able to catalyze the recombination reaction in heterologous host E. coli without a Streptomyces-encoded host factor. It is likely that the recombination reaction was stimulated by protein factor(s) provided by E. coli host.

μ1/6 integrase-directed in vivo recombination in E. coli cells. a Schematic representation of the recombination reaction between LITint-attP (4.5 kbp) and pACYCattB (4.3 kbp), and the expected product pRec (8.8 kbp). b Agarose gel electrophoretic patterns of restriction analysis: pACYCattB/XbaI (lane 1), LITint-attP/HpaI (lane 2), pRec/XbaI + HpaI (lane 3), and (lane M) DNA size marker. c Nucleotide sequence of the XbaI/HpaI fragment generated by digestion of the recombinant product pRec. The dotted and dashed sequences represent parts of pACYC184 and LITMUS 38, respectively. The boxed region corresponds to the attL sequence, and the core is underlined
In Vitro Recombination by μ1/6 Integrase
Previous experiments have shown that integrases of serine family promote in vitro recombination reaction only in the presence of a purified enzyme and substrates containing attP and attB, and do not require an accessory factor for integration [3, 28, 31]. In contrast, tyrosine integrases require also host protein factor and superhelicity of one of the two DNA substrates to promote the integration reaction in vitro [22, 26].
Integrase μ1/6 was also tested for its ability to catalyze recombination between μ1/6 attP and attB under in vitro reaction conditions. The successful recombination in vivo catalyzed by μ1/6 integrase in heterologous system suggested that some factor(s) encoded by E. coli host would also stimulate in vitro reaction. To test this, a crude extract was prepared from E. coli MC1061 cells and added to the reaction. Purified μ1/6 integrase was incubated with a supercoiled plasmid containing attP and a linear 399 bp attB in the presence or absence of E. coli crude cell extract. The reaction products were separated and analyzed by electrophoresis. Formation of the expected linear product with the anticipated size required the addition of E. coli crude cell extract (Fig. 5). The recombination reaction proceeded slowly (more than 10 h) and was less efficient when compared with previous in vitro experiments using λ integrase [23], integrase of mycobacteriophage L5 [24], or D29 [26]. All these integrases were stimulated by IHF encoded by their host organisms. The establishment of in vitro system for μ1/6 integrase demonstrated the requirements for host factor. The successful and efficient in vivo recombination in heterologous host catalyzed by μ1/6 integrase indicated the stimulatory activity of E. coli auxiliary factor(s), which can apparently substitute for streptomycete host factor. Thus, μ1/6 integrase system provides us with a potential tool for DNA engineering not only in Streptomyces species, but also in heterologous hosts.

In vitro integration assay with μ1/6 integrase. Supercoiled plasmid containing attP (pBSKattP, 3.4 kbp) was incubated with the linear 399 bp attB fragment in the absence (−) or presence (+) of crude cell extract of E. coli MC1061 and purified enzyme. Each reaction contained 0.03 pmol of pBSKattP and 0.3 pmol of attB fragment. Lane M DNA size marker, lane 1 (−) crude extract and (+) integrase (1 μg), lane 2 (+) crude extract (4 μl) and (−) integrase, lane 3 (+) crude extract (2 μl) and (+) integrase (1 μg), lane 4 (+) crude extract (4 μl) and (+) integrase (1 μg)
References
Abremski KE, Hoess RH (1992) Evidence for the second conserved arginine residue in the integrase family of recombination proteins. Protein Eng 5:87–91
Argos P, Landy A, Abremski K, Egan JB, Haggard-Ljungquist E, Hoess RH, Kah ML, Kalionis B, Narayana SV, Pierson LS III, Sternberg N, Leong JM (1986) The integrase family of site-specific recombinases: regional similarities and global diversity. EMBO J 5:433–440
Bibb LA, Hancox MI, Hatfull GF (2005) Integration and excision by the large serine recombinase φRv1 integrase. Mol Microbiol 55:1896–1910
Birnboim HC, Doly J (1979) A rapid alkaline procedure for screening recombinant plasmid DNA. Nucleic Acids Res 7:1513–1523
Bushman W, Thompson JF, Vargas L, Landy A (1985) Control of directionality in lambda site specific recombination. Science 230:906–911
Campbell A (1992) Chromosomal insertion site for phages and plasmids. J Bacteriol 174:7495–7499
Calos MP (2006) The phiC31 integrase system for gene therapy. Curr Gene Ther 6:633–645
Chen L, Woo SLC (2008) Site-specific transgene integration in the human genome catalyzed by phi BT1 phage integrase. Human Gene Ther 19:143–151
Esposito D, Scocca JJ (1997) Purification and characterization of HP1 Cox and definition of its role in controlling the direction of site-specific recombination. J Biol Chem 272:8660–8670
Farkašovská J, Vlček Č, Godány A (2003) Identification and characterization of an endolysin encoded by the Streptomyces aureofaciens phage μ1/6. Folia Microbiol 48:737–744
Farkašovská J, Klucar L, Vlček Č, Kokavec J, Godány A (2007) Complete genome sequence and analysis of the Streptomyces aureofaciens phage μ1/6. Folia Microbiol 52:347–358
Gabriel K, Schmid H, Schmidt U, Rausch H (1995) The actinophage RP3 DNA integrates site-specifically into the putative tRNAArg (AGG) gene of Streptomyces rimosus. Nucleic Acids Res 23:58–63
Godány A, Lacová B, Zelinka J (1990) The Streptomyces aureofaciens plasmid pIMB R8 and its use for shuttle vector construction. J Basic Microbiol 30:729–735
Gregory MA, Till R, Smith MCM (2003) Integration site for Streptomyces phage φBT1 and development of site-specific integrating vectors. J Bacteriol 185:5320–5323
Grindley NDF, Witeson KL, Rice PA (2006) Mechanisms of site-specific recombination. Ann Rev Biochem 75:567–605
Groth AC, Calos MP (2004) Phage integrases: biology and applications. J Mol Biol 335:667–678
Kieser T, Hopwood DA (1991) Genetic manipulation of Streptomyces: integrating vectors and gene replacement. Methods Enzymol 204:430–458
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. John Innes Centre, Norwich
Kim S, Moisoto de Vargas L, Nunes-Duby S, Landy A (1990) Mapping of a higher order protein–DNA complex: two kinds of long-range interactions in λ attL. Cell 63:773–781
Kim S, Landy A (1992) Lambda Int protein bridges between higher order complexes at two distant chromosomal loci attL and attR. Science 256:198–203
Kwon HJ, Tirumalai R, Landy A, Ellenberger T (1997) Flexibility in DNA in recombination: Structure of the lambda catalytic core. Science 276:126–131
Lee MH, Hatfull GF (1993) Mycobacteriophage L5 integrase-mediated site-specific integration in vitro. J Bacteriol 175:6836–6841
Nash HA, Robertson CA (1981) Purification and properties of the Escherichia coli protein factor required for λ integrative recombination. J Biol Chem 256:9246–9253
Nunes-Dűby SE, Kwon HJ, Tirumalai RS, Ellenberger T, Landy A (1998) Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res 26:391–406
Olivares EC, Hollis RP, Calos MP (2001) Phage R4 integrase mediates site-specific integration in human cells. Gene 278:167–176
Peña CEA, Stoner J, Hatfull GF (1998) Mycobacteriophage D29 integrase-mediated recombination: specificity of mycobacteriophage integration. Gene 225:143–151
Quenneville SP, Chapdelaine P, Rousseau J, Tremblay JP (2007) Dystrophin expression in host muscle following transplantation of muscle precursor cells modified with the phiC31 integrase. Gene Ther 14:514–522
Rashel M, Uchiyama J, Ujihara T, Takemura I, Hoshiba H, Matsuzaki S (2008) A novel site-specific recombination system derived from bacteriophage φMR11. Biochem Biophys Res Com 368:192–198
Sambrook J, Fritsch EF, Maniatis ST (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sedlmeier R, Werner T, Kieser HM, Hopwood DA, Schmieger H (1994) tRNA genes of Streptomyces lividans: new sequences and comparison of structure and organization with those of other bacteria. J Bacteriol 176:5550–5553
Thorpe HM, Smith MCM (1998) In vitro site-specific integration of bacteriophage DNA catalyzed by a recombinase of the resolvase/invertase family. Proc Natl Acad Sci USA 95:5505–5510
Thyagarajan B, Olivares EC, Hollis RP, Ginsburg DS, Calos MP (2001) Site-specific genomic integration in mammalian cells mediated by phage phiC31 integrase. Mol Cell Biol 21:3926–3934
Van Mellaert L, Mei L, Lammertyn E, Schacht S, Anné J (1998) Site-specific integration of bacteriophage VWB genome into Streptomyces venezuelae and construction of a VWB-based integrative vector. Microbiology 144:335–3351
Zhang L, Wang L, Wang J, Ou X, Zhao G, Ding X (2010) DNA cleavage is independent of synapsis during Streptomyces phage φBT1 integrase-mediated site-specific recombination. J Mol Cell Biol 2:264–275
Acknowledgments
This work was supported by the VEGA grant no 2/0140/11 of the Slovak Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Farkašovská, J., Godány, A. Analysis of the Site-Specific Integration System of the Streptomyces aureofaciens Phage μ1/6. Curr Microbiol 64, 226–233 (2012). https://doi.org/10.1007/s00284-011-0054-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-011-0054-7