
Abstract
Three molecular typing techniques were applied to assess the molecular relationships of Saccharomyces cerevisiae strains isolated from winery equipment, grapes, and spontaneous fermentation in a cellar located in “Zona Alta del Río Mendoza” (Argentina). In addition, commercial Saccharomyces strains widely used in this region were also included. Interdelta PCR typing, mtDNA restriction analysis, and microsatellite (SSR) genotyping were applied. Dendrograms were constructed based on similarity among different patterns of bands. The combination of the three techniques discriminated 34 strains among the 35 isolates. The results of this study show the complex relationships found at molecular level among the isolates that share the same ecological environment, i.e., the winemaking process. With a few exceptions, the yeast isolates were generally clustered in different ways, depending on the typing technique employed. Three clusters were conserved independently of the molecular method applied. These groups of yeasts always clustered together and had high degree of similarity. Furthermore, the dendrograms mostly showed clusters combining strains from winery and fermentation simultaneously. Most of the commercial strains included in this study were clustered separately from the other isolates analyzed, and just a few of them grouped with the strains mainly isolated from spontaneous fermentation. Only one commercial strain was clustered repetitively with a noncommercial strain isolated from spontaneous fermentation in the three dendrograms. On the other hand, this study has demonstrated the importance of selecting an appropriate molecular method according to the main objectives of the research.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The alcoholic fermentation of grape juice into wine by yeasts is an ecologically complex process [1]. Though many types of yeast species are associated with wine fermentation, Saccharomyces cerevisiae is the main species involved. Various studies on wine microbial ecology have elucidated the complex biodiversity involved in the development of the fermentation [2–7], establishing that Saccharomyces populations are integrated by multiple strains, even in inoculated fermentations. Consequently, it is important to have simple and appropriate methods that allow for discrimination at the strain level.
Numerous molecular techniques have been proposed to study Saccharomyces strain diversity, such as pulsed field electrophoresis [8], mitochondrial DNA (mtDNA) restriction analysis [9], and interdelta element PCR amplification [10, 11]. In recent years, the amplification of polymorphic microsatellite loci (SSRs, simple sequence repeats) has also been proposed as a powerful tool for S. cerevisiae strain differentiation [12–15].
Several studies have used molecular characterization of Saccharomyces to examine their biodiversity and distribution in vineyards, the geographic origin of the native isolates, and the impact of the enological practices on autochtonous ecosystems [16–18].
The “Zona Alta del Río Mendoza” (ZARM) is an important Argentinean wine region, which has optimum conditions for the growth of Malbec grapes for the production of high-quality wines. Currently, little information is available about the yeast populations involved in the winemaking process carried out in this region [5, 19]. A previous study attempted to identify the native Saccharomyces strains associated with winery equipment, grapes, and spontaneous fermentation on Malbec grape must from a cellar located in the ZARM region, during two consecutive years. It was demonstrated that a stable and resident Saccharomyces microbiota was present in the winery, which exhibited a dynamic behavior across different seasons and between years. Moreover, low occurrence of Saccharomyces on grapes and their limited participation during fermentation was also observed. Fermentations showed strain sequential substitution, and about 30–60% of yeast population at the end of fermentation were from the population already present in the winery. In addition, some commercial yeast strains were found during fermentation and on winery equipment in low percentage [5].
Although these results showed the microbial ecology of alcoholic fermentation in an industrial winery from Mendoza, there is still a lack of information on the genetic relationships among these Saccharomyces native populations.
The aim of this study was to assess the genetic relationships among Saccharomyces native population isolated from winery equipment, grapes, and spontaneous fermentation in a cellar located in “Zona Alta del Río Mendoza” (Argentina). Three molecular typing techniques (interdelta amplification analysis, mtDNA restriction analysis, and SSR genotyping) were applied for the molecular characterization of Saccharomyces isolates. The molecular analysis included the comparison of the polymorphism obtained by each method and the determination of the genetic relationship among the strains from the same ecological origin and selected commercial strains.
Materials and Methods
Yeast Strains
Saccharomyces strains were previously isolated from winery equipment and spontaneous fermentation of Malbec grape must from ZARM wine region during two consecutive years [5]. Twenty-eight previously isolated strains were selected as representatives of the diversity found on winery equipment and during fermentation (Table 1). Seven commercial strains originally isolated in France and widely used in the ZARM wine region were included in this study; they were named with an alphabetical code to preserve the company’s confidentiality.
Interdelta PCR Analysis
Total DNA extraction was performed as previously described by Hoffman and Winston [20]. Oligonucleotides primers delta 1 (5′-CAAAATTCACCTAT[A/T]TCTCA-3′) and delta 2 (5′-GTGGATTTTTATTCAACA-3′), or delta 12 (5′-TCAACAATGGAATCCCAAC-3′) and delta 21 (5′-CATCTTAACACCGTATATGA-3′) were used to amplify the total genomic DNA between the repeated interspersed delta sequences as previously described [10, 11]. PCR products were separated in 1.5% agarose gels in 0.5× TBE buffer. The molecular marker 100-bp DNA ladder (Promega, Madison, USA) was used as the molecular size standard. Electrophoresis gels were stained with ethidium bromide (5 μg ml−1), visualized by UV transillumination, and processed using the Gel Doc 1000 Video Gel Documentation System (BioRad, Richmond, USA).
Mitochondrial DNA Restriction Fragment Length Polymorphism (mtDNA RFLP)
The total DNA was extracted using the Wizard Genomic DNA extraction kit (Promega, Madison, USA) according to the manufacturer’s instructions. Digestions were performed with the restriction enzyme HinfI as previously described [9]. Restriction fragments were separated by electrophoresis in 1% agarose gels, stained, and visualized as described earlier for PCR products.
Analysis of SSR Loci
Quick preparation of DNA template for PCR was carried out as described by Jubany et al. [13]. The set of six SSR loci used in this study (Table 2) were chosen from a previous study [13], in which a large group of SSR loci were evaluated. In that study, nine SSRs were selected and six of them showed the highest discriminative power between closely related native strains. SSRs were named following the standardized criteria recently described by Jubany et al. PCR amplifications and electrophoresis were performed as described by Jubany et al. [13].
Cluster Analysis
The patterns of bands obtained after gel electrophoresis were employed for the construction of presence/absence matrix, taking into account the total number of different bands observed for each method. A total of 25 bands were obtained with improved interdelta PCR, 53 bands with mtDNA RFLP, and 82 fragments with the combination of the six SSR markers used. The Dice coefficient was calculated to estimate the similarity between the strains and a dendrogram was developed with the UPGMA method using NTSYSPC1.1 software (Exceter software, Setauket, USA). Cophenetic correlation as an estimator of goodness of fit of clustering was calculated by applying the same software package. The S. cerevisiae laboratory reference strain, S288c, was used as the out-group for the development of the dendrograms.
Results
Molecular Strain Characterization
About 28 wine Saccharomyces isolates from a commercial winery from ZARM (Argentina) and 7 commercial yeast strains were analyzed by applying three different molecular typing techniques. This group of yeasts was selected considering the results of a previous study developed in this wine region [5], in which the yeast populations analyzed were obtained from winery equipment, Malbec grapes, and musts during spontaneous fermentation. These latter samples were obtained at different stages of the process carried out in the same winery according to their own standard protocols, including the lack of inoculation with selected commercial strains, which is a current practice of this company for the production of red wines. Among the strains analyzed in this study, 13 yeasts were selected because they represent the “perennial” strains of the winery [5]. These yeasts were the isolates that were able to survive in the cellar, including the isolates found on winery equipment that were not used since the previous year, as well as those recovered from the winery in successive years. Some of these isolates were also found at the end of the spontaneous fermentation conducted in the same cellar (Table 1).
The results presented in Table 1 clearly indicate that according to the technique used, distinct levels of discrimination were obtained. Interdelta PCR with primers delta 1 and delta 2 was the least discriminative technique, distinguishing 15 patterns among the 35 strains analyzed (Table 1). When improved primers, delta 12 and delta 21, were used, the discrimination power was increased, allowing for the discrimination of 29 patterns of the Saccharomyces strains analyzed. On the other hand, the analysis of genetic variability of 35 Saccharomyces isolates using mtDNA RFLP allowed for the differentiation of 22 patterns. This technique showed a good level of polymorphism with an intermediate discriminating power, when compared with the other molecular markers used (Table 1).
The discrimination power obtained by combining the information from six SSR loci was very high, similar to that of the improved interdelta PCR (Table 1). Out of the 35 Saccharomyces isolates analyzed, 29 genotypes had a total of 87 alleles, ranging from 10 to 20 alleles and 12 to 24 genotypes per locus. However, the frequency of appearance of different alleles at each locus was very variable (Fig. 1). TG_VI and TTA_XIII showed a great discriminating power, differentiating 20 and 17 genotypes, respectively. Some alleles were detected more frequently, being more characteristic of the population under study. On the other hand, alleles found less frequently could indicate genotypes rarely detected. In summary, the combination of results obtained with the three applied molecular typing techniques allowed for the differentiation of 34 strains among the 35 isolates. Only two yeast isolates showed identical profile with all the molecular methods applied, and they could be considered as the same strain (Table 1).

Polymorphisms of alleles obtained for the six SSR loci on 28 native and 7 commercial S. cerevisiae wine strains. Alleles are expressed as + or − number of repeats relative to the sequenced (seq) strain
Molecular Relationships Among the Strains
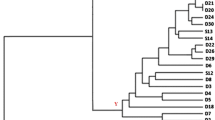
Similarities based on the Dice coefficient and UPGMA clustering from improved interdelta PCR, mtDNA RFLP, and SSR are presented in Figs. 2, 3, and 4, respectively. The dendrograms showed different degrees of relationship among the strains, according to the molecular method used. Interdelta PCR showed a superior similitude among the strains (Fig. 2), mainly because the patterns obtained by this technique have bands shared between all the isolates. The lowest genetic similitude was obtained with SSR analysis, where the first dendrogram branching was produced at a similitude coefficient value lower than 0.1. The evaluated SSR loci showed a high polymorphism between the yeast isolates analyzed, where only few of them remain closely associated (Fig. 4).

Dendrogram showing molecular relationships based on improved PCR interdelta patterns of 28 wine S. cerevisiae isolates and 7 commercial selected strains. Cophenetic correlation r = 0.88019

Dendrogram showing molecular relationships based on RFLP mtDNA patterns of 28 wine S. cerevisiae isolates and 7 commercial selected strains. Cophenetic correlation r = 0.85498

Dendrogram showing molecular relationships based on SSR analysis patterns of 28 wine S. cerevisiae isolates and 7 commercial selected strains. Cophenetic correlation r = 0.88078
The molecular methods applied reflect the polymorphism at two levels—nuclear genomic and mitochondrial genomic level. Some strains exhibited differences in each genome, by clustering together with the nuclear markers (interdelta PCR and SSR analyses) and separately with the mitochondrial marker (RFLP mtDNA), such as isolates 12, 18, and 25 (Figs. 2, 3, and 4).
With a few exceptions, the yeast isolates were generally clustered in different ways, depending on the typing technique employed. Three clusters were conserved independently of the molecular method applied (cluster 7, 9, and 24; cluster 1, 2, and 6; and cluster 14, 15, and 21) (Figs. 2, 3, and 4). These clusters were mainly integrated by isolates from the winery equipment, and mostly constituted a part of the “perennial” population of the winery. Besides these strains, the clusters included some other strains that did not remain clustered with the three molecular markers. Furthermore, the dendrograms mostly showed clusters combining strains from winery and fermentation simultaneously (Figs. 2, 3, and 4).
Most of the commercial strains included in this study were clustered separately from the other isolates analyzed, and just a few of them grouped with the strains mainly isolated from spontaneous fermentation. Only the commercial strain 35 was clustered repetitively with a noncommercial strain, grouping with strain 5 isolated from spontaneous fermentation in the three dendrograms, and showing different degrees of similarity according to the method applied (Figs. 2, 3, and 4).
Discussion
In this study, different molecular typing methods have been applied to evaluate the genetic relationships between 28 Saccharomyces strains isolated in a previous study [5]. The strains were obtained in the same cellar, and included isolates widely distributed on different winery equipment, those able to survive between vintages on the cellar surfaces, as well as those frequently found in spontaneous fermentation. Seven commercial strains widely used in this region, currently employed in this winery for the production of white wines, were also included in the analysis.
Recently, new genetic techniques have been developed to provide a differentiation of Saccharomyces strains based on single-nucleotide polymorphisms (SNPs), like multilocus sequence typing (MLS) [21, 22] or microarray techniques [23, 24]. Nevertheless, the molecular markers selected in this study still represent the simplest and most widely used techniques to study Saccharomyces biodiversity. They not only provide differentiation at the strain level but also give some information about the genetic relationship.
In the first approach, the discrimination power of the molecular techniques applied to differentiate closely related Saccharomyces strains was analyzed. Interdelta PCR with primers, delta 1 and delta 2, was the least discriminating method, as shown in previous studies [18], but the differentiation efficiency was increased with the use of improved primers [10, 25].
The combination of the allele sizes from the six SSR loci showed a high degree of resolution, even though they were not adequate for the unequivocal characterization of the present population of 35 strains. According to the observed results, the SSR technique was not equivalent to the mtDNA RFLP, and showed higher discrimination power. Furthermore, the same/different SSR amplification profile did not always corresponded to the same/different mtDNA RFLP patterns, as reported previously [18, 26]. In spite of this result, mtDNA RFLP analysis was adequate for the differentiation of yeast strains from the same ecosystem, because although some isolates were not discriminated by this technique, they remained closely related when the SSR method was used. Numerous authors have demonstrated that mtDNA RFLP analysis is an efficient technique to differentiate at the strain level [9, 18, 27, 28]. According to the results of this study, studies aiming to characterize the strains that are genetically closely related would need the inclusion of other SSR loci or the combination of SSR with other molecular typing techniques.
Molecular relationships among Saccharomyces strains from the same environment, represented by equipment, grapes, and musts spontaneously fermented from a winery located in the ZARM region, were evaluated, and complex relations were evidenced.
Only nine yeast isolates were repetitively grouped in three clusters. These groups comprised some of the “perennial” yeasts that were isolated from different winery equipment during the two vintages and were also found at the end of fermentation [5]. As shown in the dendrograms, these groups of yeasts always clustered together and had high degree of similarity. The isolates included in each of these clusters showed identical mitochondrial restriction patterns and the discrimination among them was achieved by molecular markers related to the nuclear repetitive sequences. It has been proposed that recombination processes could be favored by the presence of repeated sequences that may increase the probability of ectopic recombination events that generate different molecular patterns [29]. It is presumed that some kind of change at the nuclear level occur during the permanence of yeasts in the winery. Such changes could allow for yeast adaptation to the winery environment and survival from one vintage to the following by improving their competitive traits and tolerance to stress. Similar mechanism has been proposed by Bond to explain the dynamic genome of lager yeasts [30]. The monophyletic origin of each of these clusters and its rare relationship with the evaluated commercial strains suggest their American origin and the recent occurrence of microevolutionary events.
On the other hand, the rest of the isolates from the winery and fermentation showed a random clustering according to the molecular marker applied; this result suggests some kind of change at nuclear level, which could occur at a different rate in the nucleus with respect to the mitochondria, during the yeast life cycle. Recent studies have demonstrated a low stability of the genome in wine yeast [31], which may be due to the high reorganization capacity of its genome by Ty-promoted translocation, mitotic recombination, and gene conversion [16, 29, 32]. Alternatively, it has been proposed that ethanol and acetaldehyde introduce breaks in the DNA, with a much higher mutation rate on the mitochondrial genome. This may be due to a higher efficiency of the yeast nuclear DNA repair system compared with the mitochondrial system that lacks proofreading activity [16, 33].
In this study, the commercial strains of European origin were not restricted to a sole cluster, but were generally clustered separately or appeared related to the fermentation isolates. The existence of some genetic relation between a few “winemaking-related” strains with some of the commercial strains could support the hypothesis that some American native strains may proceed from European strains as it was recently suggested [13]. Conversely, it has been suggested that the existence of “genomic resemblance” among the native Saccharomyces and commercial strains, caused by centuries of positive selection during wine production, could lead to the same characteristics explored during the process of selection for obtaining commercial cultures [23]. These authors suggest that resemblance in phenotype is reflected in genotypic characteristics.
Results of this study show the complex relationships found at the molecular level among the yeast isolates that share the same ecological environment. Moreover, this study has revealed that beyond the abundant diversity observed, the yeasts share many genetic characteristics. On the other hand, this study has demonstrated the importance of selecting an appropriate molecular method according to the main objectives of the research.
References
Ribereau-Gayon P, Dubourdieu D, Donèche B, Lonvaud A (eds) (2006) Cytology, taxonomy and ecology of grape and wine yeast. In: Handbook of enology—the microbiology of wine and vinifications, 2nd edn, vol 1. John Wiley & Sons, Chichester, pp 1–49
Agnolucci M, Scarano S, Santoro S et al (2007) Genetic and phenotypic diversity of autochthonous Saccharomyces spp. strains associated to natural fermentation of ‘Malvasia delle Lipari’. Lett Appl Microbiol 45:657–662
Blanco P, Ramilo A, Cerdeira M, Orriols I (2006) Genetic diversity of wine Saccharomyces cerevisiae strains in an experimental winery from Galicia (NW Spain). Antoine van Leeuwenhoek 89:351–357
Lopes C, Lavalle T, Querol A, Caballero A (2005) Combined use of killer biotype and mtDNA-RFLP patterns in a Patagonian wine Saccharomyces cerevisiae diversity study. Antonie van Leeuwenhoek 3:1–10
Mercado L, Dalcero A, Masuelli R, Combina M (2007) Diversity of Saccharomyces strains on grapes and winery surfaces: Analysis of their contribution to fermentative flora of Malbec wine from Mendoza (Argentina) during two consecutive years. Food Microbiol 24:403–412
Santamaría P, Garijo P, López R et al (2005) Analysis of yeast population during spontaneous alcoholic fermentation: effect of the age of the cellar and the practice of inoculation. Int J Food Microbiol 103:49–56
Torija M, Rozes N, Poblet M et al (2001) Yeast population dynamics in spontaneous fermentations: comparison between two different wine-producing areas over a period of three years. Antonie van Leeuwenhoek 79:345–352
Blondin B, Vezinhet F (1988) Identification de souches de levures oenologiques par leurs caryotypes obtenus en électrophorèse en champ pulsé. Rev Fr Oenol 115:7–11
Querol A, Barrio F, Ramon D (1992) A comparative study of different methods of yeast strain characterization. Syst Appl Microbiol 15:439–446
Legras J, Karst F (2003) Optimisation of interdelta for Saccharomyces cerevisiae strain characterization. FEMS Microbiol Lett 221:249–255
Ness C, Lavalle F, Dubourdieu D et al (1993) Identification of yeast strains using the polymerase chain reaction. J Sci Food Agric 62:89–94
Gallego F, Perez G, Martinez I, Hidalgo P (1998) Microsatellites obtained from database sequences are useful to characterize Saccharomyces cerevisiae. Am J Enol Vitic 49:350–351
Jubany S, Tomasco I, Ponce de León I et al (2008) Toward a global database for the molecular typing of Saccharomyces cerevisiae strains. FEMS Yeast Res 8:472–484
Legras J, Ruh O, Merdinoglu D, Karst F (2005) Selection of hypervariable microsatellite loci for the characterization of Saccharomyces cerevisiae strains. Int J Food Microbiol 102:73–83
Pérez M, Gallego F, Hidalgo P (2001) Evaluation of molecular techniques for the genetic characterization of Saccharomyces cerevisiae strains. FEMS Microbiol Lett 205:375–378
Martínez C, Cosgaya P, Vásquez C et al (2007) High degree of polymorphism and geographic origin of wine yeast strains. J Appl Microbiol 103:2185–2195
Schuller D, Alves H, Dequin S, Casal M (2005) Ecological survey of Saccharomyces cerevisiae strains from vineyards in the Vinho Verde Region of Portugal. FEMS Microbiol Ecol 51:167–177
Schuller D, Valero E, Dequin S, Casal M (2004) Survey of molecular methods for the typing of wine yeast strains. FEMS Microbiol Lett 231:19–26
Combina M, Elía A, Mercado L et al (2005) Dynamics of indigenous yeast populations during spontaneous fermentation of wines from Mendoza, Argentina. Int J Food Microbiol 99:237–243
Hoffman C, Winston F (1987) A ten-minute DNA preparation from yeast efficiently release autonomous plasmids for transformation of E. coli. Gene 57:267–272
Liti G, Carter D, Moses A et al (2009) Population genomics of domestic and wild yeast. Nature 458:337–341
Vigentini I, Fracassetti D, Picozzi C, Foschino R (2009) Polymorphism of Saccharomyces cerevisiae involved in wine production. Curr Microbiol 58:211–218
Carreto L, Eiriz M, Gomes A et al (2008) Comparative genomics of wild type yeast strains unveil important genome diversity. BMC Genomics 9:524
Schacherer J, Shapiro J, Ruderfer D, Kruglyak L (2009) Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature 458:342–346
Pramateftaki PV, Lanaridis P, Typas MA (2000) Molecular identification of wine yeast at species or strain level: a case study with strains from two vine-growing areas of Greece. J Appl Microbiol 89:236–248
Schuller D, Casal M (2007) The genetic structure of fermentative vineyard-associated Saccharomyces cerevisiae populations revealed by microsatellite analysis. Antonie van Leeuwenhoek 91:137–150
Fernández-Espinar M, López V, Ramón D et al (2001) Study of the authenticity of commercial wine yeast strains by molecular techniques. Int J Food Microbiol 70:1–10
Guillamón JM, Barrio E, Querol A (1996) Characterization of wine yeast strains of the Saccharomyces genus on the basis of molecular markers: relationships between genetic distance and geographic or ecological origin. Syst Appl Microbiol 19:122–132
Puig S, Querol A, Barrio E, Pérez-Ortín JE (2000) Mitotic recombination and genetic changes in Saccharomyces cerevisiae during wine fermentation. Appl Environ Microbiol 66:2057–2061
Bond U (2009) Chapter 6 the genomes of lager yeasts. Adv Appl Microbiol 69:159–182
Pretorius IS (2000) Tailoring wine yeast for the new millenium: novel approaches to the ancient art of winemaking. Yeast 16:1–55
Rachidi N, Barre P, Blondin B (1999) Multiple Ty-mediated chromosomal translocation lead to karyotype changes in a wine strain of Saccharomyces cerevisiae. Mol Gen Genet 261:841–850
Castrejón F, Codón A, Cubero B, Benítez T (2002) Acetaldehyde and ethanol are responsible for mitochondrial DNA (mtDNA) restriction fragment length polymorphism (RFLP). Syst Appl Microbiol 25:462–467
Acknowledgments
This study was supported by Viticulture Regional Project MZASJ O7 from the Instituto Nacional de Tecnología Agropecuaria (INTA) (and Project PDT 32/06 from Dinacyt, Uruguay). L.M. is a fellow of Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and Programa de Doctorado en Ciencias Biológicas (PROBIOL), Universidad Nacional de Cuyo.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mercado, L., Jubany, S., Gaggero, C. et al. Molecular Relationships Between Saccharomyces cerevisiae Strains Involved in Winemaking from Mendoza, Argentina. Curr Microbiol 61, 506–514 (2010). https://doi.org/10.1007/s00284-010-9645-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-010-9645-y