
Abstract
A successful attempt was made to isolate linear alkylbenzene sulfonate (LAS)–degrading bacteria from soil irrigated with wastewater. The isolated bacteria were able to use LAS as sole carbon and energy source. Maximum growth rates on LAS reached only 0.27 h−1. 16S-rRNA sequencing and fatty-acid analysis placed the bacteria in the genus Enterobacter cloacae. The growth curves of E. cloacae both in the presence of and the absence of LAS were monitored using measurements of optical density at 600 nm in two different media, nutrient broth and M9 minimal medium, and were modeled mathematically. Growth in NB fit the Riccati and Voltera models, indicating that LAS is not toxic to E. cloacae cells. However, growth of E. cloacae in LAS-containing MM fit the Riccati and Voltera models, whereas growth in LAS-free MM fit the Riccati model only. Furthermore, the kinetic data shown were modeled by Monod’s, Andrew’s, and Tessier’s specific growth rate equations, coupled with the rate of consumption of different concentrations of LAS as sole carbon and energy source, and we determined that Andrew’s model best fit these data adequately as a result of the cell-inhibitory effect.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The pollution of soil and water with xenobiotics is widespread in the environment and is creating major health problems. The use of microorganisms to biodegrade organic compounds from a polluted environment represents a potential solution to such environmental problems [1]. Linear alkylbenzene sulfonate (LAS) is a widely applied anionic surfactant that accounts for approximately 25% to 30% of the world’s total synthetic surfactants. It is used in domestic as well as industrial detergent formulations and thus is discharged into wastewater collection systems worldwide [2, 3]. Commercial LAS is a mixture consisting of approximately 20 different compounds of closely related homologues and isomers, each containing an aromatic ring, sulphonated at the para-position, attached to a linear alkyl chain [4, 5].
Biodegradation is the most important mechanism for the complete removal of chemicals from the environment. It is nature’s way of getting rid of wastes by breaking down organic matter into nutrients that can be used by other organisms [6–8]. Insufficient biodegradation leads to the development of great masses of foam in streams and rivers [3, 7–9]. Therefore, all efforts must be directed to biodegrade high concentrations of LAS [3]. Many studies have been reported, but until now, no studies have confirmed the degradation of high concentrations of LAS [10]. Most research has been performed using a mixed-culture consortium.
Biodegradation of a chemical in soil is determined by its bioavailability, the presence and size of its degradation populations, and the environmental conditions that affect its activity [11]. Primary biodegradation by individual cultures and culture consortium has been established by measuring the disappearance of LAS in the culture [10, 12]. These studies showed that ≥2 cultures in a consortium complement each other as long as their combined degradation ability is much greater than that accomplished alone by individual cells, indicating catabolic cooperation between the consortium members [10]. We report the isolation and characterization of bacterial strain E. cloacae using standard methods as well as 16S-rRNA and fatty-acid analysis. This strain was capable of growing on LAS as sole carbon and energy source. A mathematic model was used to interpret the growth of this isolate on different concentrations of LAS.
Materials and Methods
Bacterial Strain Isolation
Soil samples were obtained from Al-Kherba Al-Samra, which is located near Amman, Jordan. A total of 100 g moist soil was mixed in sterile distilled water (1:1, w/v) for 1 hour at room temperature. One milliliter of that suspension was cultured into 50 ml nutrient broth (NB) or M9 minimal medium (MM) [14]. Both media were initially supplied with 50 ppm LAS and adjusted to pH 7. The growth of bacteria was checked by observing optical density at 600 nm (OD600) for 72 ± 12 hours. One-milliliter aliquots were aseptically inoculated into 50 ml MM supplemented with increasing concentrations of LAS to a maximum of 300 ppm. Growth was also checked by determining colony forming units per milliliter; this was done using dilution plate counts on nutrient agar medium. After growth had been achieved in 300 ppm LAS, selection was continued by consecutive inoculation into 50 ml MM with LAS as sole carbon and energy source. The bacterial growth was measured by spectrophotometer at OD600. The 1-ml samples of each enrichment were kept at −70°C. The growth conditions were 37°C, 150 rpm, and pH 7.4. After growth in 300 ppm LAS, the resulting population was plated onto solid MM (1.5% agar) containing 100 ppm LAS as sole carbon and energy source. The bacterial isolates that grew in 100 ppm LAS were purified; colony morphologic characteristics were noted; and biochemical identity was determined using the REMEL kit (RapID ONE and RapID NF plus systems; Remel, Lanexa, KS) procedure.
Amplification of 16S rRNA Genes
Polymerase chain reaction (PCR) was performed as previously described [15]. The 16S rRNA gene-sequencing experiments were kindly performed by the Meningitis and Special Pathogens Branch, Division of Bacterial and Mycotic Diseases, National Center for Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, GA.
Each final PCR (100 μl) contained 5 U Expand DNA polymerase (Roche, Indianapolis, IN), 2 μl DNA template, 10 mM Tris-HCl (pH 8.0), 50 mM KCl, 1.5 mM MgCl2, 200 μl (each) dATP, dCTP, dGTP, and dTTP, and 0.4 μl each eubacterial primers F8 (5′ AGTTTGATCCTGGCTCAG 3′) and R1492 (5′ ACCTTGTTACGACTT3′) [16]. Reaction mixtures were first incubated for 5 minutes at 95°C. Then 35 cycles were performed as follows: 15 seconds at 94°C, 15 seconds at 50°C, and 1 minute 30 seconds at 72°C. Reaction mixtures were then incubated at 72°C for an additional 5 minutes. PCR products were purified with a Qiaquick PCR purification kit (Qiagen, Valencia, CA).
16S rRNA Gene Sequencing
Sequencing primers were chosen from a panel of eubacterial primers: F8, R1492 (described previously), F357 (5′ TACGGGAGGCAGCAG 3′), R357 (5′ CTGCTGCCTCCCGTA3′), F530 (5′ CAGCAGCCGCGGTAATAC 3′), R530 (5′ GTATTACCGCGGCTGCTG 3′), F790 (5′ ATTAGATACCCTGGTAG 3′), R790 (5′ CTACCAGGGTATCTAAT 3′), F1068 (5′ GTCGTCAGCTCGTGTCGTGAG 3′), F1083 (5′ CGTGACATGTTGGGTTAAGTC 3′), F981 (5′ CCCGCAACGAGCGAACCC 3′), and R981 (5′ GGGTTGCGCTCGTTGCGGG 3′) [21-23]. Four additional eubacterial primers were used: R1333 (5′ CTAGCGATTCCGACTTCATGC 3′), R180 (5′ TCTCTCAAGACGTATGCGGTA 3′), R591 (5′ CATCCTGCTTAAGTAACCGTC 3′), and F1127 (5′ ATTAGTTGCCATCATTCAGTT 3′). Sequencing was performed with the Applied Biosystems (ABI, Foster City, CA) BigDye terminator cycle sequencing kit (version 2.0) according to the manufacturer’s instructions, with the exception of using 0.25 μl BigDye instead of 8 μl. Sequencing products were purified using Centri-Sep spin columns (Princeton Separations, Adelphia, NJ), and the sequencing products were resolved with an ABI model 3100 automated DNA sequencing system (ABI).
Evaluation of the 16SrRNA Gene Sequences of Enterobacter spp by Computer Analysis
16S rRNA gene sequencing of the amplified products, which was obtained with PCR primers F8 and R1492, generated 929-bp–long sequences that had 98.5% identity to the sequence of previously published Enterobacter spp. 16S rRNA gene sequences and 98.1% identity match to various Klebsiella pneumoniae sequence entries. The software used for all data analysis was GCG Wisconsin, version 10.2 (Accelrys, San Diego, CA). The raw trace files from the ABI 3100 sequencer were visually examined, aligned, and edited (SEQMERGE). E. cloacae sequences were compared using DISTANCES to determine the level of similarity among them. The sequences made in this study were compared with previously published Enterobacter spp. and Klebsiella pneumoniae sequences by using BESTFIT to determine levels of identity between sequences.
Fatty-acid Analysis
Fatty-acid analysis experiments were kindly conducted by T. L. Pitt at the Laboratory of HealthCare Associated Infection, Microbiology Division London, UK. Fatty-acid analysis (MIDI-Sherlock) of cell extracts results placed the organism in the genus Enterobacter with the highest match to E. cloacae. Therefore, the bacterial strain isolated was considered to be E. cloacae
Growth Conditions
In all experiments conducted, LAS-containing uninoculated media served as control. NB and agar medium were used for isolation of bacteria and in some degradation tests performed. MM was used to verify the degradability of LAS by the bacteria used in the study. MM as described by Miller [14], consisted of the following: 3.0 g Na2HPO4, 1.5 g KH2PO4, 1.0 g NH4Cl, and 0.5 g NaCl dissolved in 500 mL distilled water with pH adjusted to 7.4 with 6 N NaOH. Then the following compounds were added: 0.24 g MgSO4, 0.05 g CaCl2 · 6H2O, and 0.05 g FeCl3 · 6H2O. After autoclaving, the following were added by predissolving each in 2 mL distilled water and sterilizing by filtration through a 0.45-μm Millipore filter: 0.001 g thiamine-HCl. LAS (300 ppm) was used as carbon and energy source. The initial cell mass (starting inocula) for each experiment for the NB and MM media experiments were grown from single colonies in two consecutive shake flask cultures (overnight followed by 3-hour recovery) in NB. Cells from 3-hour cultures were harvested by centrifugation and then washed with and resuspended in NB at a concentration of 0.5 OD600 units. One milliliter of these bacterial cells was used to inoculate 50 ml of either media in 125-ml Erlenmeyer flasks for shake-flask experiments. Samples were then taken at time intervals and assayed for cell mass at optical density at OD600. Growth curves using OD600 and viable counts for specific growth rates calculations were performed as previously described [3, 13].
Analytic Methods
LAS was analyzed in culture media using the classic methylene blue method [17], modified by Li and Rosen [18], as previously described [3]. The disappearance of LAS was qualified by reverse-phase high-performance liquid chromatography (HPLC) on a Spherisorb ODS2 column (250 × 4.6 mm; 5-μm particle size; motile phase 0.11 M NaClO4 with acetonitrile gradient [0% to 100% vol/vol]), and compounds were detected spectrophotometrically at 255 nm [3].
Results and Discussion
Bacterial Identification
Using 16S rRNA gene sequencing, a 929-bp sequence was recovered. BLAST analysis indicated a 98.5% identity match to various Enterobacter spp. sequences and a 98.1% identity match to various K. pneumonia sequence entries. However, fatty-acid analysis placed the strain in the genus Enterobacter with the highest match to E. cloacae. Based on these procedures, along with the API system results, we concurred that the bacterial strain belongs to E. cloacae. To our knowledge, ours is the first study of LAS biodegradation by E. cloacae. In fact, although microbial degradation of LAS compounds has been discussed widely in the literature, no studies involving E. cloacae have been published.
Effect of Substrate Concentration on LAS Biodegradation by E. cloacae
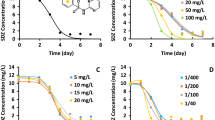
Different substrate concentrations were used to investigate their effect on the biodegradation level of LAS (Fig. 1a). It was apparent that ≥300 ppm LAS resulted in <60% biodegradation. As a result, an inverse relation between the extent of LAS concentration and extent of biodegradation was shown. Decreased LAS degradation at higher concentrations was unlinked to a toxicity effect on the bacterial culture as predicted from the Andrews model in Eq. 7 and as reported with white rot fungus Phanerochaete chrysosporium [18]. To determine the exact time point for achieving complete degradation of 100 ppm LAS (Fig. 1b), LAS biodegradation was assessed at 2-hour time point intervals. It was found that complete degradation of 100 ppm LAS was achieved within 18 hours (Fig. 2b) compared with the results of a previous study showing degradation as occurring within days [8]. The 300-ppm LAS concentration resulted in the highest rate of LAS degradation (6.25 ppm/h).

Effect of different substrate concentration on the biodegradation of LAS by E. cloacae. (a) data collected at 24 h intervals, (b) data collected every 2 h intervals. The figure represents the percentage of remaining LAS expressed as active matter under the same experimental conditions (37°C, pH 7.4 and agitation rate of 150 rpm). All data are average of three trials with error bars indicating STDEVs (σn−1)

HPLC chromatograms showing the finger print of: (a) standard solution of LAS (100 ppm), (b) complete degradation of LAS (100 ppm)
Growth Curves and Mathematic Interpretation
E. cloacae cells were grown in NB and in NB plus 300 ppm LAS (Fig. 3a and b). In the second culture (MM), LAS was used as sole carbon and energy source (Fig. 4 and Table 2). Cells grown in LAS-containing NB had a shorter lag phase than that observed in cells grown in LAS-free NB media. Any growth of E. cloacae cells in LAS-containing MM was a function of exhaustion of LAS substrate because no other carbon source existed in MM, thus confirming LAS biodegradation as established by HPLC results (Fig. 2). Any further possibility for elimination of this anionic surfactant from the culture by surface adsorption or extensive chemical degradation is doubtful [3]. Moreover, the lag phase for cells grown in MM was shorter than the same counterpart cells grown in NB. It has been reported that single bacterial isolates are known to have limited ability for degrading the alkyl chain, and they also lack the capability to cleave the sulfonated aromatic ring that exists in the LAS aromatic ring [3, 8].

Growth curve of E. cloacae on (a) nutrient broth media (LAS-free NB) and (b) LAS-containing NB medium. The curve represents the absorbance at 600 nm as a function of time. Data were fitted by Riccati and Volterra models; this was done under the same experimental conditions (37°C, pH 7.4 and agitation rate of 150 rpm)

Growth curve for E. cloacae grown on LAS-free minimal medium (M9) and 300-ppm LAS-containing M9 minimal medium. Growth was conducted under 37°C, pH 7.4 and agitation rate of 150 rpm
The inhibitory influence of substrate has been described by two types of growth kinetics, the Riccati and Voltera models (Fig. 3a and b). This growth was modeled based on increasing cell mass with time in the presence and the absence of population history (OD600 measurements). The Riccati model assumes that the inhibition of the cells is directly proportional to the square of population growth according to Eq. 1:
where β is a constant related to the percentage of inhibition, and x is the cell mass. This Riccati equation can be integrated to give the logistic curve according to Eq. 2:
The logistic curve predicted in Eq. 2 shows a sigmoidal profile, which leads to a stationary population of size xs = 1/β. In contrast, the Volterra model is used to predict a phase of decrease after the stationary population has exhausted all viable resources. It can be described thusly in Eq. 3:
where K o is a constant account for the history or memory of population. The sign, K o , is taken as negative for compounds that inhibit growth and positive for compounds that promote growth. The cell populations in LAS-free NB (Fig. 3a) and in LAS-containing NB (Fig. 3b) were the same, indicating that LAS had less toxicity on these cells. Both the Riccati and Voltera models clearly fit the experimental data adequately. The corresponding model parameters are listed in Table 1. Similar results were obtained when comparing these results with cells grown in MM. However, the Riccati model showed the best fit to the experimental data compared with the Voltera model, especially for the MM control (i.e., LAS-free MM) (Table 2). Experimental data for the growth of cells in MM plus 300 ppm LAS showed an adequate fit for both models. It is obvious from these experimental data that cells have little memory effect on growth, indicating that they can resist the toxicity of the LAS. Kramer et al. [20] found that E. cloacae tolerate up to 25% sodium dodecyl sulfate rather than metabolize it.
Toxicity of LAS Toward Bacterial Cells
A series of batch experiments were conducted based on viable cell number (Fig. 5) to determine toxicity of E. cloacae toward LAS. In these experiments, initial LAS concentrations varied from 500 to 3,000 ppm. Taking into account the effect of these concentrations on cell number, the specific growth rate, μ, can be determined experimentally by determining the ratio of the difference in cell concentration to difference in time [21] according to Eq. 4:

The relationship between substrate concentration and growth rate, this carried out by three different models, all the models could fit the experimental data (Monod, Tessier and Andrews) being Andrews the best fit, this was done under the same experimental conditions (37°C, pH 7.4 and agitation rate of 150 rpm)
The Monod, Tessier, and Andrews models describe the correlation between substrate concentration and specific growth rate as long as saturation kinetics for cellular systems can be formed. The Monod model has the following form (Eq. 5):
where μmax is the maximum specific growth rate when S ≫ Ks. More related forms of specific growth rate dependence have been proposed, which, in particular instances, give better fits to experimental data. For example, Tessier and Andrews, respectively, suggest the following models (Eqs. 6 and 7):
The specific growth rate may be inhibited by medium constituents, such as substrate or product. This inhibition effect may be predicted by Eq. 7:
The specific growth curve that describes the relation between substrate concentration and growth rate for cells is shown in Fig. 5. The inhibition constant for the substrate (Table 3) is relatively high (1600 mg/l) which is a measure of sensitivity to inhibition by LAS indicate that the inhibition by LAS is negligible. Moreover, all of the models could fit these data. The corresponding parameters are listed in Table 3. Decreased growth rate may have been caused by the production of several inhibitory metabolic intermediates or by decreasing bioavailability of LAS to bacterial cells with increasing LAS concentration. This bacterial isolate is thus interesting, and distinct model systems for the study of LAS biodegradation and kinetics could be useful. Finally, E. cloacae may be helpful in the bioremediation of LAS-contaminated soil and water.
References
Widada J, Nojiri H, Omori T (2002) Recent developments in molecular techniques for identification and monitoring of xenobiotic-degrading bacteria and their catabolic genes in bioremediation. Appl Microbiol Biotechnol 60:45–59
Larson RJ, Rothgeb TM, Shimp RJ, Ward TE, Ventullo RM (1993) Kinetics and significance of biodegradation of alkylbenzene sulfonate in the environment. J Am Oil Chem 70:645–657
Khleifat K (2006) Biodegradation of linear alkylbenzene sulfonate by a two-member facultative anaerobic bacterial consortium. Enzyme Microb Technol 3:1030–1035
Olmo M, Garballo A, Nimer M, Lopez I, Ferrer J, Vtichez J (2004) Development of methodologies for different degrees of resolution of linear alkylbenzene sulfonates in groundwater and wastewater by liquid chromatography using sodium dodecyl sulfate. Chromatographia 60:157–164
García MT, Campos E, Ribosa I, Latorre A, Sánchez-Leal J (2005) Anaerobic digestion of linear alkyl benzene sulfonates: biodegradation kinetics and metabolite analysis. Chemosphere 60:1636–1643
Kloepper-Sams P, Torfs F, Feijtel T, Gooch J (1996) Effects assessments for surfactants in sludge-amended soils: a literature review and perspectives for TRA. Sci Total Environ 185:171–185
Rapaport RA, Eckhoff WS (1990) Monitoring linear alkyl benzene sulfonate in the environment: 1973–1986. Environ Toxicol Chem 9:1245–1257
Jiménez L, Breen A, Thomas N, Federle TW, Sayler GS (1991) Mineralization of linear alkylbenzene sulfonate by a four-member aerobic bacterial consortium. Appl Environ Microbiol 57:1566–1569
Jakobi G, Löhr A (1987) Detergent ingredients. In: Gerhartz W (ed) Ullmann’s encyclopedia of industrial chemistry, 5th edn. VCH, Weinheim, Gremany, pp 338–372
Schleheck D (2003) Biodegradation of synthetics surfactants: linear alkylbenzene sulfonates (LAS) and related compounds. PhD Thesis, University of Konstanz, Germany
Knaebel DB, Federle TW, McAvoy DC, Vestals JR (1994) Effect of mineral and organic soil constituents on microbial mineralization of organic compounds in a natural soil. Appl Environ Microbiol 60:4500–4508
Swisher RD (1987) Surfactant biodegradation, 2nd edn. New York. NY, Dekker, pp 587–614
Khleifat K (2006) Biodegradation of phenol by Ewingella americana: effect of carbon starvation and some growth conditions. Process Biochem 41:2010–2016
Miller JH (1972) Experimental in molecular genetics. Cold Spring Harbor, NY
Gee JE, De BK, Levett PN, Whitney AM, Novak RT, Popovic T (2004) Use of 16S rRNA gen sequencing for rapid confirmatory identification of Brucella isolates. J. Clin Microbiol 42:3649–3654
Eden PA, Schmidt TM, Blakemore RP, Pace NR (1991) Phylogenetic analysis of Aquaspirillum magnetotacticum using polymerase chain reaction-amplified 16SS rRN-specific DNA. Int J Syst Bacteriol 41:324–325
Longwell J, Maniece WD (1955) Determination of anionic detergent in sewage effluents and river water. Analyst 80:167–171
Li Z, Rosen M (1981) Two-phase mixed indicator titration method for determination of anionic surfactants. Anal Chem J 53:1516–1519
Yadav JS, Lawrence DL, Nuck BA, Federal TW, Reddy CA (2001) Biotransformation of linear alkylbenzene sulfonated (LAS) by Phanerochaete chrysosporium: oxidation of alkyl side-chain. Biodegradation 12:443–453
Kramer VC, Calabrese DM, Nickerson KW (1980) Growth of Enterobacter cloacae in the presence of 25% sodium dodecyl sulfate. Appl Environ Microbiol 40:973–976
Nuhoglu A, Yalcin B (2004) Modeling of phenol removal in a batch reactor. Process Biochem 40:1233–1239
Acknowledgment
We would like to thank Professor Jay E. Gee from the Division of Bacterial and Mycotic Diseases, National Center for Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, GA, USA for his help in the identification of the bacterial strain used in this study and proofreading the Materials and Methods section.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Khleifat, K.M., Tarawneh, K.A., Ali Wedyan, M. et al. Growth Kinetics and Toxicity of Enterobacter cloacae Grown on Linear Alkylbenzene Sulfonate as Sole Carbon Source. Curr Microbiol 57, 364–370 (2008). https://doi.org/10.1007/s00284-008-9203-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-008-9203-z