
Abstract
Legionellaceae is a family of Gram-negative, mesophilic, and facultative intracellular parasitic bacteria that inhabits freshwater environments. In this article, the Legionella population of water samples from the North and South Lake, located close to the Brazilian Scientific Station on King George Island, Keller Peninsula, Antarctica has been characterized. Culture onto selective medium and a independent-culture method were applied to the samples. In our attempt to isolate Legionella species from Antarctic lakes, we were able to obtain one L. pneumophila colony by an amoebic coculture procedure followed by plate culture onto a selective medium. In addition, results obtained from phylogenetic inference showed the presence of noncharacterized specimens of Legionella spp. These findings indicated the presence of legionellae in Antarctica and suggest that these bacteria can adapt to extreme conditions and open new possibilities for understanding the survival strategies of mesophilic Legionellaceae living in Antarctic environments. Furthermore, the isolation of these symbiotic bacteria in Antarctic lakes will allow future studies on cold-resistant mechanisms of legionellae in polar environments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Legionella species might exist as different forms in the environment: for example, free-living planktonic cells, intracellular parasites of free-living protozoa, inhabitants of mixed-community biofilms, and viable but nonculturable bacteria [16, 20]. Legionellae concentration in aquatic environments is usually low [9]. However, this microorganism is widespread in freshwater habitats and its ubiquity is probably due to the bacteria’s capacity to survive in a wide range of environmental conditions (i.e., temperature, pH, and salinity [6, 9, 23]). Furthermore, the ability of Legionella pneumophila to survive and grow in a cold environment and the mechanisms of bacterial resistance at a lower temperature has been reported [19, 24]. Recently, molecular characterization of a Legionella population confirmed the occurrence of this bacterium as a member of the microbial community of the aquatic environment, even at low temperatures [26].
In the current study, both culturing and culture-independent methods were used to isolate and characterize the Legionella species in freshwater lakes of Keller Peninsula, Antarctica. The purpose of this study was to study the occurrence and genetic diversity of Legionellaceae bacteria within the microbiota of Antarctic lakes.
Materials and Methods
Sampling and Concentration of Water
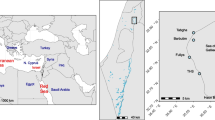
Water samples were collected from two lakes on the Keller Peninsula of King George Island, Antarctica. The lakes are located at S 62°5′1.8″/W 58°23′36.16″ (North Lake) and S 62°5′9.0″/W 58°23′39.75″ (South Lake), near the Brazilian Scientific Station “Comandante Ferraz” (BSSCF). Both lakes are pristine and used for the water supply of BSSCF. The temperature of water at the time of sampling measured 0°C in both lakes. The samples were collected on December 2004 at the beginning of the Antarctic summer. The samples, 5 L each, were collected in sterile plastic flasks and filtered through a sterile polycarbonate membrane (47 mm in diameter, pore size of 0.45 μm, Millipore, Ireland) at the BSSCF laboratory. Each membrane filter was aseptically removed from the support grid and transferred to a sterile 50-mL polypropylene centrifuge tube with 10 mL of the original water sample. The cells were released from the membrane by vortexing the tubes for 5 min. Aliquots of concentrated water samples were kept frozen, at −20°C, during transport to Brazil for subsequent molecular analyses.
Cultivation of Environmental Isolates
Concentrated water samples (0.1 mL) were inoculated in triplicate on buffered charcoal yeast extract agar supplemented with glycine, vancomycin, and polymyxin B (αBCYE-GVP) as previously described [6]. The culture procedure of environmental samples was done at BSSCF within 2 h after environmental sampling and concentration procedures, and the plates were incubated for 10 days in a humidified atmosphere at 37°C. Presumptive legionellae colonies were subcultured onto αBCYE-GVP and αBCYE agar in the absence of cysteine. Colonies that grew on αBCYE-GVP but were unable to grow on αBCYE without cysteine were considered putative Legionella strains and were checked for autofluorescence under ultraviolet light (365 nm), Gram stained, and subcultured again onto a new selective medium. As this method failed to subculture any strains, we enriched Legionella environmental isolates obtained from the primary plating culture method with an amoebic coculture, as described by Moffat and Tompkins [17]. Axenic cultures of Acanthamoeba castellanii ATCC 30011 strains (American Type Culture Collection, Rockville, MD) were used in the amoebic coculture procedure. Briefly, A. castellanii trophozoites were grown in a 25-cm2 cell culture flask (Costar, Cambridge, MA, USA) containing Neff’s amoebic medium (120.0 mg NaCl, 3.0 mg MgCl2. 6H2O, 3.0 mg CaCl2, 3.0 mg FeSO4, 142.0 mg Na2HPO4, 136.0 mg KH2PO4, 10.0 g proteose peptone, 18.0 g glucose, 1.0 L distilled water, pH 6.8). Amoebas were collected for the coculture procedure by vigorous agitation of the media when cells had formed a confluent layer on the bottom of the flask (1.0 × 106 cells/mL). Amoebas were harvested and pelleted by centrifugation at 1000g for 10 min. The supernatant was removed, and the amoebas were resuspended in 20 mL of PAS buffer (Page’s amoebic saline; 120 mg NaCl, 4 mg MgSO4 · 7H2O, 4 mg CaCl2 · 2H2O, 142 mg Na2HPO4, 136 mg KH2PO4, 1.0 L distilled water). The washing procedure was repeated and the amoebas were resuspended in 10 mL of PAS, at a final concentration of 2.0 × 105 cells/mL. Finally, aliquots of 1 mL of amoebic suspension were transferred to into 24-well cell culture microplates (Costar, Cambridge, MA, USA) and allowed to adhere to the wells for 1 h at 37°C before inoculating putative legionellae colonies. An aliquot of 100 μL of each amoebic suspension was cultured in triplicate onto sheep blood agar before amoebic coculture procedures in order to check for the absence of exogenous contamination, and L. pneumophila ATCC 33152 strains was used as a positive control. In the amoebic coculture procedure, 100 μL of each suspension was inoculated in duplicate on both selective (αBCYE-GVP) and nonselective (αBCYE without cysteine) media agar plates by the spread plate technique. Plates were incubated at 37°C and observed for 10 days to determine the presence of intracellular bacteria.
DNA Isolation from Cultures and Environmental Samples
Genomic DNA from both legionellae putative colonies and concentrated water samples (2 mL) was extracted with the QIAamp DNA kit (Qiagen, Hilden), eluted in 50 μL of AE buffer (supplied in the kit) and stored at −20°C. MilliQ ultrapure water was used as a negative control. The concentration of DNA was determined by electrophoresis on 0.8% agarose gels.
PCR Amplification and Sequencing of Environmental Isolates
Preliminary identification of isolates was based on the nearly complete 16S rRNA and specific defect of organelle trafficking A (dotA) gene sequences. Reagents concentration and thermal cycling conditions used in the polymerase chain reactions (PCRs) followed previous descriptions [3, 4, 11, 13, 18], as shown in Table 1. Oligonucleotide primers targeting regions of each of the genes were used to amplify 430- to 1525-bp products encompassing regions of variation (Table 1). A previous comprehensive new database was carried out in order to check their specificity and, consequently, to prevent false-positive results. PCR products to be sequenced were purified with the PureLink PCR Purification Kit (Invitrogen Life Technologies, California, USA). The sequencing reaction was done according to the MegaBACE 1000 protocol (Amersham Biosciences, Piscataway, NJ). Both strands of each amplicon were sequenced. Lane tracking and base calling were done with Cimarron 3.12 software.
Molecular Cloning Library and Sequencing
The PCR products (430 pb) generated from genomic DNA extracted from freshwater samples were subsequently cloned into pGEM-T® Vector System I (Promega Corporation, Madison, WI, USA) according to manufacturer’s instructions. Plasmid DNA from the selected colonies was extracted as described previously [2]. Cloned inserts were reamplified with primers LEG-448 and LEG-858. The purity and concentration of each PCR product was verified on 1% agarose gels.
Phylogenetic Analyses
The 16S rRNA gene sequences from both environmental isolates and clones were analyzed and concatenated with the BioEdit Sequence Alignment Editor version 5.0.9 [10]. The nucleotide sequences obtained were compared to the GenBank data library with BLAST software [1]. The possible chimerical sequences were checked using the CHECK-CHIMERA tool [15]. Phylogenetic and molecular evolutionary analyses from 16S rRNA and dotA gene sequences were done with MEGA software version 3.1 [12], and phylogenetic trees were constructed with the neighbor-joining (NJ) method without gaps.
Nucleotide Sequence Accession Numbers
The 16S rRNA gene sequences from the cloning library chosen for this study were deposited in the GenBank database under accession Nos. DQ404565 to DQ404588. Partial 16S rRNA gene sequences from environmental strains were deposited under accession Nos. DQ646380 and DQ646381. Sequences from 16S rRNA gene obtained with eubacterial universal primers were deposited under accession Nos. DQ646382 and DQ646383, whereas dotA gene sequences from environmental strains were deposited under accession Nos. DQ646378 and DQ646379.
Results
Preliminary Isolation of Legionella spp. from Water Samples
A single suspected Legionella colony was observed on a selective culture medium inoculated with water samples from North Lake and grew poorly when subcultured after coculturing with A. castellanii. In addition, a single colony was observed on the αBCYE-GVP agar medium when duplicates where plated. Neither isolate was autofluorescent under ultraviolet light. This colony failed to grow on αBCYE agar without l-cysteine, suggesting that the Legionella specimen was isolated from freshwater of North Lake.
PCR from Isolates and Genomic DNA Extracted from Environmental Samples
The PCR amplification followed by sequencing of the 16S rRNA gene from an environmental isolate DNA template by using both eubacterial universal primers (27F/1525R and 338F/1100R) and primer sets that target the Legionella 16S rRNA gene (LEG-448/ LEG-858 internal primers) showed the presence of Legionella and Yersinia bacteria in the single colony obtained by the coculture amoebic procedure. For this reason, a virulence-related dotA gene from the same DNA template was amplified and sequenced in order to identify both Legionella and Yersinia species. Phylogenetic relationships of putative Legionella colonies obtained in this study, designated Strain-03 and Strain-04, and L. pneumophila, inferred from nucleotide sequences of partial dotA and 16S rRNA genes, are shown in Figs. 1 and 2, respectively. Sequence identity analyses to both partial dotA and 16S rRNA genes from Strain-03 and Strain-04 nucleotide sequences showed the same homology of 98.8% to dotA (GenBank accession No. AF095231) and 16S rRNA (GenBank accession No. AE017354) gene sequences of L. pneumophila subsp. pneumophila strain Philadelphia 1. In contrast, 16S rRNA gene sequences obtained by amplifying and sequencing the same DNA template with eubacterial primers from Strain-03 (1472-bp fragment length) and Strain-04 (1474-bp fragment length) showed 99.8% and 97.8% identities with Y. kristensenii and Y. aldovae, respectively.

Phylogenetic relationships of L. pneumophila inferred from partial dotA gene sequences (428-bp) of environmental strains obtained in this study (designed Strain-03 and Strain-04) and GenBank database. A midpoint rooting option was applied to root the tree due to the absence of a reliable outgroup. The bootstrap values presenting corresponding branches were evaluated from 1000 replications. Values below 75% are not indicated. The scale bar represents 0.2 substitutions per 100 nucleotides

Bootstrap consensus tree of partial 16S rRNA gene (420-bp) generated by LEG-specific primers (LEG-448 and LEG-858). Comparative analysis were done between putative Legionella strains, designated as Strain-03 and Strain-04, and clone sequences from DNA extracted directly from North Lake water, clones designated as LN, and South Lake water, clones designated as LS. The neighbor-joining (NJ) method was used as distance matrix analysis and Tamura–Nei correction was used to compensate for different evolutionary rates in both NJ phylogenetic trees. The 16S rRNA gene sequence of Coxiella burnetii was used as outgroup to root tree. Branches supported by values higher than 50% in the bootstrap analysis (1000 replications) are indicated in nodes. The bars represent 2% estimated sequence divergence
Molecular Cloning Library of 16S rRNA Gene of Antarctic Lakes
LEG-specific primers (LEG-448 and LEG-858) were used to construct cloning libraries of 16S rRNA gene sequences. The inserts containing a fragment of 16S rRNA gene (430 bp) from 24 clones—15 clones from South Lake (LS) and 9 clones from North Lake (LN)—were sequenced in both strands. The primer sequences on both sides of the sequence were removed, and 417 bp from each clone sequence were considered for alignment. Analyses of clone sequences obtained from the 16S rRNA gene matched the GenBank sequence database of L. pneumophila, L. lytica, L. jeonii, L. erythra, and Legionella-like amoebal pathogens (strain LLAP-10). Phylogenetic analyses revealed that clone sequences from both North Lake and South Lake samples clustered into three main groups (Fig. 2). One of these groups consisted of the following clones: LNcl04, LScl03, LScl04, LScl07, LScl11, LScl19, LScl21, and LScl22 (similarity above 98% and bootstrap values of 54–91%), which formed a common lineage with L. pneumophila. Clones LNcl01, LNcl02, LNcl03, LNcl09, LNcl10, and LScl16, grouped together with an unidentified Legionella specimen designed LLAP-10 (similarities above 95%). Finally, the third main group was represented by clones LNcl06, LNcl07, LScl01, LScl02, LScl05, LScl06, LScl10, and LScl18, which grouped together with L. lytica. Other clones of legionellae belonged to L. jeonii, which showed the closest relation to the clone LNcl11 (similarity 94.4%), whereas clone LScl14 was related to L. erythra (similarity 94.4%).
Discussion
In this study, the importance of natural hosts in the process of resistance and the development of Legionella bacteria at low temperatures was shown when the L. pneumophila strain was first obtained by the amoebic coculture procedure. Although the interaction between Y. aldovae and free-living protozoa has not been described in the literature, the presence of the Enterobacteriaceae family genera has been reported in extreme temperature conditions from polar waters [8]. These symbiotic bacteria could inhibit growth or mask the presence of Legionellaceae colonies on selective agar media [14]. Moreover, sequencing analysis of the dotA gene was consistent with results previously obtained by using internal primers of the 16S rRNA gene. In this specific case, the choice of the dotA gene for confirming the L. pneumophila isolates was based on the fact that this gene is a component of the pathogenicity island, which contains the 24 dot/icm genes on 2 unlinked 22-kb regions on the L. pneumophila genome [11]. This gene has also been related to adaptation and intracellular survival of this bacterium in different hosts, such as amoeba, ciliates, and other free-living protozoans [25].
The amplification of Yersinia specimen 16S rRNA gene sequence by eubacterial universal primers could be explained by different DNA concentrations between the Legionella and Yersinia strains within a mixture culture. The PCR products containing both Legionella and Yersinia amplicons allowed for identifying Y. aldovae or Y. kristensenii, which could be present at a higher DNA concentration, by preferentially amplifying the PCR reaction. Consequently, the sequencing reaction of the V3 region to the 16S rRNA gene allowed only the Yersinia specimen to be identified when eubacterial universal primers were used. In contrast, when an internal fragment of the 16S rRNA gene that encoded to Legionella species was amplified and sequenced with LEG-448 and LEG-858 primers, L. pneumophila was likely to be present at lower levels. Preferential amplification is more likely in mixed-target PCR reactions where competition between multiple targets for primer annealing could result in the numerically dominant sequences being preferentially amplified [7]. An additional hypothesis could be related to the specificity of primers used: LEG-448 and LEG-858 primers were designed from the V3 region of the 16S rRNA gene molecule for specific detection of Legionella strains, whereas the universal primers were designed to bind to nearly all 16S rRNA gene sequences of the eubacteria. Therefore, results obtained could be mainly influenced not only by different DNA concentrations but also by the specificity of primers used.
The concentration of metallic ions, mainly Al, Fe, and Cu, in frozen lakes and the metal requirements for growth of Legionellaceae could represent a potential environmental precondition for survival of these microorganisms in Antarctic aquatic systems. In the present study, water samples were collected at the beginning of the Antarctic summer, when the surface ice begins to shrink and the air and water temperatures are considerably lower. Cowan and Tow [5] showed that during summer, peripheral moats (melting of the peripheral ice cover) allow the inflow of water from the melting streams, resulting in steep salt gradients in the water column. In addition, Schaefer et al. [22] reported the chemical composition and the concentration of metallic ions, such as iron (0.050 mg/mL, North Lake, and 0.06 mg/mL, South Lake) and zinc (0.525 mg/mL, North Lake, and 0.853 mg/mL, South Lake), in water from the same lakes sampled in this study.
In conclusion, Antarctic lakes could provide a buffered and protected aquatic environment for Legionellae, where the population of these bacteria is phylogenetically diverse. Sequencing results of 16S rRNA gene partial fragments showed that most of Legionellae clone sequences were related to the presence of protozoa hosts and that the population of Legionella bacteria in both lakes is phylogenetically diverse. The symbiotic association of Legionella species and amoebas could allow for the mechanisms of survival and resistance of the bacteria at low-temperatures environments. Moreover, our results indicated the presence of uncommon and protozoonotic Legionella species in a polar environment. Although the process of obtaining viable Legionellae under extreme conditions (i.e., low temperatures) would be difficult and time-consuming, it could explain certain factors that are not well understood, such as adaptation to the cold and changes in the cellular structure that could have occurred in an evolutionary stage.
References
Altschul SF, Madden TL, Schäffer AA et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acid Res 25:3389–3402
Ahn SC, Baek BS, Oh T et al (2000) Rapid mini-scale plasmid isolation for DNA sequencing and restriction mapping. Biotechniques 29:466–468
Brosius J, Palmer ML, Kennedy PJ et al (1978) Complete nucleotide sequence of a 16S ribosomal RNA gene from Escherichia coli. Proc Natl Acad Sci USA 75:4801–4805
Brosius J, Dull TL, Sleeter DD et al (1981) Gene organization and primary structure of a ribosomal RNA operon from Escherichia coli. J Mol Biol 140:107–127
Cowan DA, Tow LA (2004) Endangered Antarctic environments. Annu Rev Microbiol 58:649–690
Carvalho FRS, Vazoller RF, Foronda AS et al (2007) Phylogenetic study of Legionella species in pristine and polluted aquatic samples from a Tropical Atlantic Forest ecosystem. Curr Microbiol 55:288–293
Dahllöf I, Baillie H, Kjlleberg S (2000) rpoB-Based microbial community analysis avoids limitations inherent in 16S rRNA gene intraspecies heterogeneity. Appl Environ Microbiol 66:3376–3380
Dancer SJ, Shears P, Platt DJ (1997) Isolation and characterization of coliforms from glacial ice and water in Canada’s High Arctic. J Appl Microbiol 82:597–609
Fliermans CB, Cherry WB, Orrison LH et al (1981) Ecological distribution of Legionella pneumophila. Appl Environ Microbiol 41:9–16
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Ko KS, Hong SK, Lee HK et al (2003) Molecular evolution of the dotA gene in Legionella pneumophila. J Bacteriol 185:6269–6277
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, New York, NY, pp 115–175
Lye D, Fout GS, Crout SR et al (1997) Survey of ground, surface, and potable waters from the presence of Legionella species by EnviroAmp PCR Legionella kit, culture, and immunofluorescent staining. Water Res 31:287–293
Maidak BL, Cole JR, Lilburn TG et al (2000) The RDP (Ribosomal Data Project) continues. Nucleic Acids Res 28:173–174
Mampel J, Spirig T, Weber SS et al (2006) Planktonic replication is essential for biofilm formation by Legionella pneumophila in a complex medium under static and dynamic flow conditions. Appl Environ Microbiol 72:2885–2895
Moffat JF, Tompkins LS (1992) A quantitative model of intracellular growth of Legionella pneumophila in Acanthamoeba castellanii. Infect Immun 60:296–301
Miyamoto H, Yamamoto H, Arima K et al (1997) Development of a new seminested PCR method for detection of Legionella species and its application to surveillance of legionellae in hospital cooling tower water. Appl Environ Microbiol 63:2489–2494
Pasko-Kolva C, Shahamat M, Colwell RR (1993) Effect of temperature on survival of Legionella pneumophila in the aquatic environment. Microb Releases 2:73–79
Piao Z, Sze CC, Barysheva O et al (2006) Temperature-regulated formation of mycelial mat-like biofilms by Legionella pneumophila. Appl Environ Microbiol 72:1613–1622
Säwström C, Anesio MA, Granéli W et al (2006) Seasonal viral loop dynamics in two large ultraoligotrophic Antarctic freshwater lakes. Microb Ecol 53:1–11
Schaefer C, Francelino MR, Simas FNB et al (2004) Ecossistemas Costeiros e Monitoramento Ambiental da Antártica Marinha. NEPUT, Viçosa, Minas Gerais, Brazil
Sheehan KB, Henson JM, Ferris MJ (2005) Legionella species diversity in an acidic biofilm community in Yellowstone National Park. Appl Environ Microbiol 71:507–511
Söderberg MA, Rossier O, Cianciotto NP (2004) The type II protein secretion system of Legionella pneumophila promotes growth at low-temperatures. J Bacteriol 186:3712–3720
Swanson MS, Hammer BK (2000) Legionella pneumophila pathogenesis: a fateful journey from amoebae to macrophage. Annu Rev Microbiol 54:567–613
Wullings BA, van der Kooij D (2006) Occurrence and genetic diversity of uncultured Legionella spp. in drinking water treated at temperatures below 15°C. Appl Environ Microbiol 72:157–166
Acknowledgments
This work was supported by The National Council for Scientific and Technological Development (CNPq) and Brazilian Antarctic Program (PROANTAR). F.R.S.C. was supported by a doctoral fellowship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Carvalho, F.R.S., Nastasi, F.R., Gamba, R.C. et al. Occurrence and Diversity of Legionellaceae in Polar Lakes of the Antarctic Peninsula. Curr Microbiol 57, 294–300 (2008). https://doi.org/10.1007/s00284-008-9192-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-008-9192-y