
Abstract
A mature and diverse T and B cell receptor repertoire is a prerequisite for immunocompetence. In light of its increased susceptibility to infection, the human fetus has long been considered deficient in this regard. However, data accumulated since the 1990s and in earnest in the past couple of years paints a more complicated picture. As we describe in this review, mechanisms responsible for generating a diverse receptor repertoire, such as somatic recombination, class switch recombination, and somatic hypermutation, are all operational to surprising extents in the growing fetus. The composition of the fetal repertoire differs from that of adults, with preferential usage of certain variable (V), diversity (D), and joining (J) gene segments and a shorter complementarity determining (CDR3) region, primarily due to decreased terminal deoxynucleotidyl transferase (TdT) expression. Both T and B cell receptor repertoires are extremely diverse by the end of the second trimester, and in the case of T cells, are capable of responding to an invading pathogen with in utero clonal expansion. Thus, it would appear as though the T and B cell receptor repertoires are not a hindrance towards immunocompetence of the newborn. Our improved understanding of fetal receptor repertoire development is already bearing fruit in the early diagnosis of primary immunodeficiencies (PID) and may help clarify the pathogenesis of congenital infections, recurrent abortions, and autoimmune disorders in the near future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The adaptive immune system develops in the growing fetus under usually sterile conditions. Unlike most organ systems, and similar to the brain, the immune system is not fully formed at birth, and it continues to expand and mature after it is introduced to the outside world, well into early infancy [1]. Maternal immune protection, in the form of trans-placental and breast milk antibody transfer, buys the fetus (and neonate) time before it must fend for itself [2]. As this issue of Seminars in Immunopathology focuses on the immunocompetence of the newborn, the question we wish to answer here is—could the fetus, stripped of this protective maternal cape, develop an independent immune response to an infecting agent?
To do so, fetal T and B cells would first need to recognize the invading pathogen—a task which requires a diverse repertoire of specific antigen receptors, so that by chance, one (or more) of these receptors recognizes an invading antigen. These fetal B and T cell receptor (BCR and TCR, respectively) repertoires are the focus of this review, as we attempt to map out the order and fashion in which they develop throughout gestation, to assess whether they reach a threshold of maturity we may describe as immunocompetence. TCR and BCR repertoire development plays a significant role in a wide range of pathological conditions. Underdevelopment of the fetal immune system can result in primary immunodeficiencies or insufficient response to pathogens in congenital or perinatal infections. Altered development may result in autoimmune disorders or malignancies and may be a factor in recurrent abortions [3, 4]. Understanding normal T and B cell receptor repertoire development could therefore have translational applications in detecting, preventing, or treating these pathologies.
Due to constraints of both space and scope, the focus of this review will be placed solely on “classic” adaptive immunity, i.e., B cells and αβT cells, and will not attempt to cover γδT cell and Natural Killer cell fetal development.
T and B cell receptor assembly and repertoire diversification
During T and B cell development, a mechanism termed DNA rearrangements creates a unique TCR or immunoglobulin (Ig) sequence, encoding for a unique antigen receptor, in each individual T and B cell, respectively. This mechanism was first elucidated in the mid 1970’s by Susumu Tonegawa, who correctly postulated that an advanced combinatorial interplay within these loci is required to produce the incredible diversity of TCRs and Igs [5]. Lymphocyte receptors differ from one another primarily in the third complementarity determining region (CDR3) of their variable domains. In the pre-combined germline Ig and TCR loci, the CDR3 contains multiple iterations of variable (V), joining (J), and in the immunoglobulin heavy chain and TCR β chain, diversity (D) gene segments. During somatic DNA rearrangements, lineage specific recombination machinery selects V, D, and J segments, excises the “unchosen” segments from the genome, and links the selected segments to form a unique VDJ sequence for each lymphocyte (Fig. 1a). Additional diversity is obtained through random insertion and deletion of nucleotides in the junctions between rearranged segments, including the potential addition of palindromic (P) nucleotides, and by the terminal deoxynucleotidyl transferase (TdT)—catalyzed addition of non-germline encoded (N) nucleotides [6]. Diversity is than further multiplied by the joining of the two rearranged chains that make up the receptor (heavy and light chains in Ig, α, and β in TCR). In B cells, avidity maturation processes following antigen stimulation (class switch recombination, somatic hypermutation) increase total diversity even further.

a An illustration of somatic recombination with D-J rearrangement followed by V-DJ rearrangement. During recombination, an excision circle is formed, unique to the selected VDJ combination. b Upon excision of the T cell receptor δ during T cell receptor α rearrangement, a ubiquitous T cell excision circle (TREC) is formed, representative of most αβT rearrangements. Quantitative PCR quantification of TREC serves as a surrogate for VDJ recombination measurement
Assessing receptor repertoire maturity and diversity
Immunocompetence, definitive though it may sound, is a spectrum. To determine immunocompetence, it does not suffice to verify its existence but also to quantify it. A lymphocyte receptor repertoire may be mature (having undergone somatic recombination) but not diverse. It may be diverse, but abnormal. It may be diverse and normal but occur in only a handful of B or T cells. To adequately quantify the immunocompetence of the fetal lymphocyte receptor repertoire, one needs to find how many rearranged receptors there are, the diversity of their receptor repertoire, and its composition.
T and B cell receptor assembly
Quantifying the process of somatic recombination presents a unique challenge. Because of the enormous diversity of VDJ recombination events, no single gene segment can be used as a marker to assess overall production of all rearranged B or T cell receptors. During rearrangement, the gene segments excised from the chromosome (“unchosen” V, D, and J segments) remain within the cell as episomal gene circles, termed excision circles. For each VDJ combination, a different excision circle is formed, and so, like their chosen counterparts, most excision circles cannot be used as a single marker to assess overall rearrangement (Fig. 1a). However, in αβT cells, a common requirement for productive rearrangement of the TCRA locus is deletion of the TCRD locus which it encompasses. The excised segment of TCRD, from the δ-rec locus to the ψ-Jα locus, forms a ubiquitous excision circle shared by most rearranged αβT cells (Fig. 1b). Quantification of this excision circle, termed T cell receptor excision circle (TREC), reflects production of rearranged αβT cells at large [7]. A similar byproduct of BCR rearrangement, Kappa-deleting receptor excision circle (KREC), is used to quantify VDJ recombination in B cells [8].
Excision circles are readily quantifiable with real-time PCR and have become staples of adaptive immune system assessment in immunology labs worldwide. TREC measurement at birth is currently used for severe combined immunodeficiency (SCID) newborn screening in several countries, and its use is expanding annually [9]. Its accuracy allows for early diagnosis and initiation of protective measurements or treatment for SCID and other severe forms of lymphopenia. Both TREC and KREC are used routinely in the assessment of immune reconstitution after bone marrow transplant [10], and in the evaluation of primary and secondary immunodeficiencies [11].
Repertoire analysis
Receptor repertoire diversity reflects how many different receptors exist and how evenly they are distributed within the repertoire. The healthy receptor repertoire is diverse and evenly distributed (polyclonal), whereas in pathological conditions such as acute infection or primary immunodeficiencies (Omenn’s syndrome, for instance), the repertoire is restricted and dominated by one (clonal) or several (oligoclonal) expanded clones. Techniques for measuring this diversity have evolved over the years, from targeted amplification and sequencing of selected rearranged products [12], through detection of different V gene segments with fluorescent monoclonal antibodies [13], to next-generation sequencing (NGS) of the entire receptor repertoire [14].
The most readily available tool for repertoire assessment for both research and diagnostic purposes is measurement of receptor expression by flow cytometry. Representatives of specific TCR Vβ families are detected and quantified using flow cytometry and compared to a large cohort of healthy controls. Over or under-expression of specific Vβ families compared to the control group can indicate pathologies, such as immunodeficiency, autoimmunity, or infection (Fig. 2a). For evaluation of junctional diversity, the P and N indels between the selected VDJ segments, a technique termed spectratyping is often used. In a diverse repertoire, a Gaussian distribution of CDR3 lengths will exist for each V-J combination. In a clonal setting, the dominant clone will peak above the rest. For spectratyping, multiplex PCR with fluorescent-labeled specific primers is performed, and PCR products are analyzed by separation of the various fragments according to their lengths (Fig. 2b). In recent years, NGS has overtaken all other forms of receptor repertoire analysis for research purposes, due to the wealth of information it provides for each sample analyzed. In immunosequencing, the CDR3 regions of all T or B cells within a sample are sequenced in parallel. The sequences are then annotated for their V, D, and J segments, and the content of their junctions are mapped out as well. This allows for both tremendous volume (as many as hundreds of thousands of sequences per sample) and depth of data (Fig. 3). Unlike other methods of diversity assessment, immunosequencing allows us to measure the contributions of both combinatorial (VDJ selection) and junctional (P and N) diversity generating mechanisms. It also enables calculating repertoire diversity with different diversity indices, in order to compare the diversity of different samples (fetuses of different gestational ages, healthy, and ill).

Commonly used receptor repertoire assessment methodologies. a T cell receptor Vβ (TCRVβ) expression via flow cytometry—fluorescent-labeled monoclonal antibodies for 24 TCRVβ families are used to detect the relative expression of the different Vβ families on the entire T cell population in a sample. The results are then compared to a reference card of healthy adult controls (white bars). A 26-gestational-week-old fetus (black bars) displays a diverse, normal usage pattern compared to controls. b Spectratyping—complementarity determining region 3 (CDR3) length distribution for a specific V-J pairing. Gaussian distribution reflects normal junctional diversity

Tree map representation of T (a) and B cell (b) receptor repertoires from a 12- (left) and a 26-gestational-week-old fetus (right), where each square represents a unique CDR3 sequence, and the size of the square represents its relative frequency within the receptor repertoire [14]
Fetal B cell receptor repertoire development
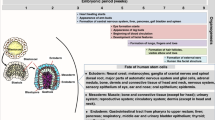
When does rearrangement of B cell receptors begin? Nickerson et al. first reported rearranged Ig heavy chain (IGH) transcripts in fetal bone marrow and liver as early as 9 weeks gestation [15]. In a concerted effort with Lee and Notarangelo, we found and quantified KREC, as a surrogate of rearranged IGH, in fetal blood from 12 weeks gestation on. The process of VDJ recombination in fetal B cells accelerates logarithmically throughout gestation, culminating in thousands of KREC copies in samples from third trimester fetuses (Fig. 4) [14].

Summary of fetal T and B cell receptor repertoire developmental landmarks
In terms of their composition, fetal B cells appear capable, by mid-second trimester, of expressing all V, D, and J gene segments. However, we and others, most notably Schroeder and Zemlin, whose contributions to the field are innumerable, have detected significant variations between fetal and adult repertoire composition.
The fetal B cell receptor repertoire is characterized by shorter average CDR3 length with decreased N diversity compared to children and adults [14, 16, 17]. Rother et al. observed decreased expression of both DNTT, which encodes TdT itself, as well XRCC4, a gene encoding a protein by the same name known to form a complex with TdT, shown to promote N-nucleotide additions [18]. In the same study, Rother et al. link between IL-7 receptor alpha (IL7RA) expression, which is also decreased in the fetus, and TdT expression. We and others have demonstrated incremental increases in N-nucleotide, and subsequently CDR3 length, along with gestational age.
As shown by Schroeder and others, IGH VDJ usage pattern in the fetus also differs from that of adults, with preferential usage of DH-proximal V, JH-proximal D, and DH-proximal J gene segments, as well as preferential usage of DH7-27 [12, 14, 17, 19, 20]. The long standing assumption that proximity of rearranged segments dictates this preference due to immaturity of the fetal recombination machinery has never been proven [18]. Another theory raised by Schroeder, who observed that the overexpression of certain gene segments such as VH3 is conserved among several species, is that certain gene segments encode for receptors which provide survival benefits and were selected for by evolutionary pressures [12].
Next-generation sequencing of the BCR repertoire suggests that as early as 12 weeks gestation, the receptor repertoire is already fairly diverse and evenly distributed, substantially more so than its TCR counterpart (Fig. 3). By late second trimester, the fetal BCR repertoire is as diverse as that of a healthy infant [14].
The study of affinity maturation in-utero has only begun to scratch the surface. We and others have shown that fetal B cells undergo class switch recombination, primarily to IgG, as early as late second trimester [14, 21]. A small number of IgA transcripts were found in one fetal blood sample taken in early third trimester [14]. Griffiths et al. found IgM in the cord blood of 83/93 fetuses infected with congenital cytomegalovirus (CMV) infection. IgM was absent in all 104 cord blood samples from non-infected infants [22]. This would suggest that while B cells are capable of producing low levels of antibodies in response to congenital infection, they appear not to actively produce antibodies in utero unstimulated. With Lee and Notarangelo, we have detected somatic hypermutation (SHM) of Ig transcripts in B cell receptors from samples as young as 12 weeks gestation, in the earliest samples examined for it [14]. SHM is non-random, reflecting antigen-selection, in all samples examined, but in particular in late second trimester and onwards. Pascual and Zemlin have shown that while the rate of SHM increases throughout gestation, it remains well short of adult levels at birth [19, 21].
With the capacity to produce a large number of rearranged B cells and with a diverse receptor repertoire capable of affinity maturation, B cell incompetence in the fetus appears to be more regulatory than maturational.
Fetal T cell receptor repertoire development
While the humoral branch of the fetal (and neonatal) adaptive immune system is allowed to develop in the shadow of maternal antibodies, fetal T cells are left to fend for themselves. And yet, despite its outsized role in fetal immunocompetence, the fetal αβT cell receptor repertoire has been the subject of far less examination than that of its B cell counterpart.
George and Schroeder found rearranged TCR β chain (TRB) DJ transcripts in human fetal thymus tissue as early as 8 weeks gestation [23]. Raaphorst et al. demonstrated fully rearranged TRB VDJ transcripts in fetal liver, spleen, and blood from as early as 11 weeks gestation [24]. In all tissues examined, VDJ usage in early fetal life was significantly restricted.
TREC is present for the first time in fetal blood only around week 13 of gestation, as evidenced by six fetal blood samples dated week 12 gestation for which no TREC copies were detectable with RT-PCR. Thus, TCR recombination appears to lag behind its BCR counterpart by approximately 1 week. Like KREC, TREC rises logarithmically throughout gestation, and by mid-second trimester, it reaches levels that would be considered immunocompetent for a healthy child [14].
The fetal TRB repertoire appears to be more substantially restricted in early pregnancy than that of IGH. Fetal TRB displays a more limited and skewed VDJ usage pattern than IGH. In 14- and 15-week-old fetal livers, Raaphorst et al. demonstrated several absent TRBV and TRAV gene families [13]. We found similar results in the blood of a 14-week-old fetus, with absent TRBV5-5 and TRBV18 receptors on fetal T cells. TRB usage, however, becomes more similar to adult usage as pregnancy progresses, from 20 weeks gestation on (Fig. 2a) [14]. All fetal TRBV repertoires examined by us display overexpression of TRBV5-6, even after the fetal TRB usage pattern as a whole normalizes, evoking Schroeder’s hypothesis of evolutionary benefit. George and Schroeder found very low levels of TdT expression in an 8-week-old fetal thymus, with higher expression levels in 11- and 16-week-old thymi, but still considerably lower than in adult thymus. As in B cells, low TdT expression in T cells manifests in minimal N additions in early T cell development compared to adults, leading to a shortened CDR3 overall [23, 25]. Our results validate these findings and indicate incremental increases in N-nucleotide additions and CDR3 length as pregnancy progresses [14]. Gavin and Bevan demonstrated that T cell clones from the TCR repertoire of TdT-knockout mice are less specific than clones from wild-type mice. They speculate that the promiscuity afforded by fewer N-nucleotide additions compensates for decreased diversity in allowing a limited TCR repertoire to recognize a greater variety of antigens [26].
In terms of diversity, the fetal TCR repertoire is significantly more restricted and uneven than the BCR repertoire in early second trimester fetuses, as evident by immunosequencing (Fig. 3). By the beginning of the third trimester, however, TRB repertoire diversity reaches a level equivalent to that of a healthy infant [14].
Taken together, evidence regarding T cell receptor repertoire development suggests that while it lags behind B cell receptor development, by mid-gestation, the fetal TRB repertoire is diverse and mature enough to recognize an invading pathogen.
Furthermore, studies into the fetal response to viral infection, primarily CMV, have shown that fetal T cell can not only recognize, but respond to an invading pathogen. Miles et al. isolated CMV-specific CD8+ T cells from the cord blood of infants with congenital CMV infection using MHC class I tetramers. Virus specific T cells underwent clonal expansion, acquired a differentiated phenotype, and were capable of producing perforin, granzyme A, and pro-inflammatory cytokines, although interferon-γ production was limited in young infants [27–29]. An important observation from these studies is that congenital infection may affect not only the antigen specific, responding T cells, but the remainder of the T cell receptor repertoire as well [27]. Alternatively, in the cord blood in infants of hepatitis C virus (HCV) infected mothers, Babik et al. found T cells to be relatively suppressed, in both their differentiation status and cytokine release profiles [30]. As HCV is far less likely to cause congenital infection, the differing responses to congenital CMV (activation) and HCV (suppression) infections could be seen as measured fetal immune responses to threats of varying degrees [28, 30].
Conclusions and future prospects
It has long been known that lymphocytes are the dominant leukocyte in fetal blood, comprising close to 90% of all white blood cells in blood from second and third trimester fetuses [31]. The maturity and functionality of these abundant lymphocytes, however, has only recently been clarified and remains a subject of intense research. Development of the human fetal T and B cell receptor repertoires begins somewhere towards the end of the first trimester, with small numbers of rearranged lymphocytes making their way from the fetal thymus, bone marrow, and liver towards the blood (Fig. 4). This primordial adaptive immune system is immature, with a restricted, skewed repertoire mostly lacking in advanced maturation features (N-nucleotide additions, class switch recombination, and somatic hypermutation). By the end of the second trimester, however, T and B cell receptor repertoires make great strides in their maturity level, reaching diversity measurements equivalent or near-equivalent to those of postnatal infants.
The consequences of this developmental leap are manifold. The difference between first and second trimester antigen receptor maturation levels could account for the very different outcomes seen in first trimester congenital infections compared to late pregnancy infections. Our understanding of fetal immune maturity could help dictate ideal time-windows for vaccination, intra-uterine interventions, and diagnostic evaluation of early onset immune system pathologies.
One clinical application of our insights into normal T cell receptor repertoire development has already emerged. As newborn screening for SCID with TREC measurement is implemented in more and more countries with each passing year [9], the subgroup of preterm infants detected through the newborn screening program poses a diagnostic dilemma [32]. Should a 26-gestational-week-old preterm infant with a low TREC copy number be suspected of SCID, or does his low TREC measurement simply reflect normal development? Using this newfound knowledge bank of fetal TREC values and repertoire diversity, true SCID patients can successfully be filtered out from false positive, healthy infants.
In conclusion, while the research field of fetal antigen receptor repertoires is still in its infancy, a general outline has begun to form. Additional studies are required, in particular regarding the shape of these repertoires under varying pathological conditions. Whereas many nuances remain to be elucidated, it is reasonable to determine that the fetal T and B cell receptor repertoires are not a hindrance towards acquiring immunocompetence.
References
Marchant A, Kollmann TR (2015) Understanding the ontogeny of the immune system to promote immune-mediated health for life. Front Immunol 6(77):1–3
Niewiesk S (2014) Maternal antibodies: clinical significance, mechanism of interference with immune responses, and possible vaccination strategies. Front Immunol 5(446):1–15
Leveque L, Khosrotehrani K (2014) Feto-maternal allo-immunity, regulatory T cells and predisposition to auto-immunity. Does it all start in utero? Chimerism 5(2):59–62
Taub JW, Ge Y (2004) The prenatal origin of childhood acute lymphoblastic leukemia. Leuk Lymphoma 45(1):19–25
Tonegawa S, Steinberg C, Bernardinj A (1974) Evidence for somatic generation of antibody diversity. Proc Natl Acad Sci 71(10):4027–4031
Zemlin M, Schelonka RL, Bauer K, Schroeder HW (2002) Regulation and chance in the ontogeny of B and T cell. Immunol Res 26:265–278
Douek DC et al (1998) Changes in thymic function with age and during the treatment of HIV infection. Nature 396(6712):690–695
Langerak AW et al (2004) Unraveling the consecutive recombination events in the human IGK locus. J Immunol 173:3878–3888
Kwan A, Puck JM (2015) History and current status of newborn screening for severe combined immunodeficiency. Semin Perinatol 39(3):194–205
Hochberg EP et al (2001) Quantitation of T-cell neogenesis in vivo after allogeneic bone marrow transplantation in adults. Blood 98(4):1116–1121
Amariglio N, Lev A, Simon AJ et al (2010) Molecular assessment of thymus capabilities in the evaluation of T-cell immunodeficiency. Pediatr Res 67(2):211–216
Schroeder HW, Wang J (1990) Preferential utilization of conserved immunoglobulin heavy chain variable gene segments during human fetal life. Proc Natl Acad Sci 87:6146–6150
Raaphorst FM et al (1994) Usage of TCRAV and TCRBV gene families in human fetal and adult TCR rearrangements. Immunogenetics 39(5):343–350
Rechavi E, Lev A, Lee YN et al (2015) Timely and spatially regulated maturation of B and T cell repertoire during human fetal development. Sci Transl Med 7(276):1–12
Nickerson KG, Berman J, Glickman E, Chess L, Alt F (1989) Early human IgH gene assembly in Epstein-Barr virus-transformed fetal B cell lines. J Exp Med 169:1391–1403
Souto-Carneiro MM, Sims GP, Girschik H, Lee J, Lipsky PE (2005) Developmental changes in the human heavy chain CDR3. J Immunol 175(11):7425–7436
Zemlin M et al (2001) The diversity of rearranged immunoglobulin heavy chain variable region genes in peripheral blood B cells of preterm infants is restricted by short third complementarity-determining regions but not by limited gene segment usage. Blood 97(5):1511–1513
Rother MB et al (2016) Decreased IL7Rα and TdT expression underlie the skewed immunoglobulin repertoire of human B-cell precursors from fetal origin. Sci Rep 6:33924. doi:10.1038/srep33924
Pascual V, Verkruyse L, Casey ML, Capra JD (1993) Analysis of IgH chain gene segment utilization in human fetal liver. J Immunol 151:4164–4172
Schroeder HW, Zhang L, Philips JB (2001) Slow, programmed maturation of the immunoglobulin HCDR3 repertoire during the third trimester of fetal life. Blood 98(9):2745–2751
Zemlin M et al (2007) The postnatal maturation of the immunoglobulin heavy chain IgG repertoire in human preterm neonates is slower than in term neonates. J Immunol 178(2):1180–1188
Griffiths PD, Stagno S, Pass RF, Smith RJ, Alford CA Jr (1982) Congenital cytomegalovirus infection: diagnostic and prognostic significance of the detection of specific immunoglobulin M antibodies in cord serum. Pediatrics 69(5):544–549
George JF, Schroeder HW (1992) Developmental regulation of D beta reading frame and junctional diversity in T cell receptor-beta transcripts from human thymus. J Immunol 148(4):1230–1239
Raaphorst FM, Kaijzel EL, van Tol MJ, Vossen JM, van den Elsen PJ (1994) Non-random employment of V beta 6 and J beta gene elements and conserved amino acid usage profiles in CDR3 regions of human fetal and adult TCR beta chain rearrangements. Int Immunol 6(1):1–9
Prabakaran P et al (2012) Expressed antibody repertoires in human cord blood cells: 454 sequencing and IMGT/HighV-QUEST analysis of germline gene usage, junctional diversity, and somatic mutations. Immunogenetics 64(5):337–350
Gavin MA, Bevan MJ (1995) Increased peptide promiscuity provides a rationale for the lack of N regions in the neonatal T cell repertoire. Immunity 3(6):793–800
Miles DJC et al (2007) Cytomegalovirus infection in Gambian infants leads to profound CD8 T-cell differentiation. J Virol 81(11):5766–5776
Marchant A et al (2003) Mature CD8+ T lymphocyte response to viral infection during fetal life. J Clin Invest 111(11):1747–1755
Huygens A et al. (2015) Functional exhaustion limits CD4+ and CD8+ T-cell responses to congenital cytomegalovirus infection J Infect Dis 212(3):484–494.
Babik JM, Cohan D, Monto A, Hartigan-O’Connor DJ, McCune JM (2011) The human fetal immune response to hepatitis C virus exposure in utero. J Infect Dis 203(2):196–206
Forestier F, Daffos F, Catherine N, Renard M, Andreux JP (1991) Developmental hematopoiesis in normal human fetal blood. Blood 77(11):2360–2363
Barbaro M et al (2017) Newborn screening for severe primary immunodeficiency diseases in Sweden—a 2-year pilot TREC and KREC screening study. J Clin Immunol 37(1):51–60
Acknowledgments
Raz Somech is supported by the Jeffrey Modell Foundation (JMF). This review and some of the work were performed in partial fulfillment of the requirements of the PhD of Erez Rechavi at the Sackler School of Medicine (Tel Aviv University).
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Immunocompetence of the Newborn -- Guest Editors: Arnaud Marchant and Tobias Kollmann
Rights and permissions
About this article

Cite this article
Rechavi, E., Somech, R. Survival of the fetus: fetal B and T cell receptor repertoire development. Semin Immunopathol 39, 577–583 (2017). https://doi.org/10.1007/s00281-017-0626-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-017-0626-0