
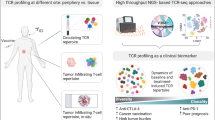
Abstract
Characterizing the interaction of cancer cells with the host adaptive immune system is critical for understanding tumor immunology and the modus operandi of immunotherapeutic interventions to treat cancer. As the key cellular effectors of adaptive immunity, T cells are endowed with specialized receptors (the T cell receptor; TCR), to recognize and to eliminate cancer cells. The diversity of the TCR repertoire results from specialized genetic diversification mechanisms that generate an incredible variability allowing recognizing extensive collections of antigens. Based on the attainment and function of the TCR, the TCR repertoire is a mirror of the human immune response, and the dynamic changes of its usage can be assumed as a promising biomarker to monitor immunomodulatory therapies. Recent advances in multiplexed PCR amplification and massive parallel sequencing technologies have facilitated the characterization of TCR repertoires at high resolution even when only biomaterial of limited quantity and quality, such as formalin-fixed paraffin-embedded (FFPE) archived tissues, is available. Here, we review the concept framework and current experimental approaches to characterize the TCR repertoire usage in cancer including inherent technical and biological challenges.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recent years brought a large number of approvals of immunomodulatory therapies for the treatment of cancer. Most of these therapies are immune checkpoint inhibitors targeting the CTLA-4 or PD-1 pathways. Notably, the first reports to successfully treat cancer patients by activating the immune system date back more than 100 years. In 1868, Busch intentionally infected a cancer patient with erisypelas leading to tumor shrinkage [1]. At that time, the causative agent of erisypelas had not been identified. Hence, when 25 years later Coley injected “Coley’s toxin”, a mixture of heat-inactivated Streptococcus pyogenes and Bacillus prodigiosus, into cancer patients, this can be regarded as the first defined immunotherapeutic approach [2]. In 1909, Paul Ehrlich proposed that tumor formation is normally suppressed by the immune system [3], a concept elaborated by Burnet as tumor immunosurveillance [4].
Tumor immunosurveillance
Each day, humans come in contact with millions of potential pathogens. Due to the immune system with the different defense mechanisms, these foreign encounters normally take a non-hazardous cause. Besides pathogens, however, cells can also impose a thread to the organisms. In this regard, in each cell, thousands of DNA lesions occur per day. Most of these are corrected by different DNA repair pathways for various types of DNA damage. Indeed, only 1 in 1000 accidental base changes results in a permanent mutation [5], which might result in malignant transformation of the cell. Such potentially dangerous cells have to be eliminated or at least controlled. According to the immunosurveillance theory, this task is executed by the immune system. Currently, direct proof of immunological destruction of premalignant lesions in vivo is lacking. Several observations, however, indicate the possibility of immunological control of tumors. Indeed, an impaired immune system by primary immunodeficiency, immunosuppressive drugs, or human immunodeficiency virus infection is associated with increased cancer risk [6–9]. Accordingly, organ transplants from donors with previous history of malignancies might develop transmitted tumors in the organ recipient, even when the initial donor’s tumor was more than 30 years ago curatively resected [10, 11]. This long-lasting control of tumor growth by immune cells was nicely demonstrated in a sarcoma mouse model: treatment of mice with a low dose of carcinogen caused an initial growth of tumor which was subsequently stabilized over 150 days. Upon depletion of CD4+ and CD8+ T cells, however, tumors started to grow again [12]. In the clinical setting, immunological restraint of tumor progression can be deduced from the association of quantity, type, and localization of immune cells in primary tumors and clinical outcome; for most tumor entities, a brisk, intratumoral infiltrate of cytotoxic T-lymphocytes (CTL) predicts a better prognosis (reviewed in [13]). In addition, the most frequently proposed mechanism of spontaneous regression of cancer was host immunity, where other postulated factors like infection or postoperative trauma might also influence the immune response [14].
Adaptive immunity and tumor antigens
NK cells are regarded as the first line of defense against premalignant cells. Indeed, when cells accumulate mutations, different cellular programs are initiated, rendering the cell immunogenic. For example, DNA damage response can activate p53 leading to cell cycle arrest and activation of the senescence program. Different cytokines secreted by these cells recruit and enhance the activity of NK cells. In addition, oncogenic stress induces the expression of NK cell-activating ligands. Especially, NKG2D ligands expressed on the premalignant/malignant cells can trigger their destruction by NK cells [15]. In a study of single nucleotide polymorphisms in the natural killer complex, different haplotypes were identified associated with natural cytotoxic activity. These SNPs were almost all located in the NKG2D gene, and individuals with a haplotype associated with decreased natural cytotoxicity had an increased risk of cancer development [16]. Similarly, in NKG2D-deficient mice, spontaneous tumor developed faster than in mice with normal NKG2D expression [17]. Control of transformed cells by NK cells is probably restricted to early lesions. Indeed, in the earlier described sarcoma mouse model, depletion of NK cells had no effect on tumor growth, i.e., tumors remained in their dormant state [12]. In humans, NK cell activity can be impaired by the tumor by inducing expression of inhibitory receptors and downregulation of activating receptors by NK cells [18]. In addition, in established tumors, the frequency of NK-cell can be decreased compared to adjacent tumor tissue [19, 20]. A change in the frequency of NK-cell infiltration thus might be a promising approach to treat cancer. This hypothesis is sustained by mouse models; when tumor-bearing mice got the opportunity to train by wheel running, this exercise led to distinct reduction of tumor incidence and growth. As underlying mechanism, a significant increase of NK-cell infiltration into the tumor by mobilizing the cells through epinephrine was identified [21].
The innate immune response is not only able to attack the tumor, but proper innate immune signaling is required for induction of an adaptive immune response. Indeed, molecular analyses of T cell-infiltrated tumors revealed a type I interferon signature indicative of innate immune response [22]. Subsequent studies in mice revealed that a type I response was required for spontaneous T cell priming [23]. The IFN secretion occurs mainly by dendritic cells (DCs), requires cytosolic tumor-derived DNA sensing through the STING pathway [24], and leads to cross-priming of CD8+ T cells by CD8α+ DCs [25]. In 1989, the first murine tumor antigen, a mutated self-protein, recognized by CTL was identified revealing how transformed cells can be distinguished from normal cells [26]. Shortly thereafter, the first human tumor antigen, a peptide derived from MAGE1, was reported [27]. Thus, CTL can recognize peptides of different size with the majority of natural presented peptides being nine amino acids long presented by MHC class I molecules [28]. These peptides are derived from intracellular proteins and can be classified as (i) overexpressed, (ii) differentiation, (iii) cancer-germline, (iv) mutated, or (v) viral antigens. Overexpressed or differentiation antigens are characterized by low tumor specificity since their expression is also present in the corresponding tissue or in normal cells and are thus referred to tumor-associated antigens (TAA). Cancer-germline antigens (CTA) are expressed in a wide variety of cancer types due to the general demethylation present in some cancers including their promoter region. Although these proteins are also expressed by male germline cells and trophoblasts, they can be considered as tumor-specific since the respective cells do not express MHC class I molecules. The both remaining classes, i.e., viral and mutated antigens, compose the tumor-specific antigens (TSA). Viral proteins are only expressed in infected cells or tumor cells in cancers with viral etiology. Mutated proteins in tumor cells can give rise to neoantigens either by converting a non-binding into a binding peptide or changing the MHC class I presented peptide sequence. Notably, the majority of mutations do not translate into neoantigens recognizable by autologous T cells, and the majority of neoantigens are derived from passenger mutations [29, 30]. Neoantigen-based immunotherapies will therefore require a personalized medicine approach.
One major difference between the different classes of antigens is the T cell receptor (TCR) affinities. During development, the fate of a T cell is determined by the interaction of its TCR with self-peptide–MHC complexes that are displayed by thymic antigen-presenting cells. Only when the TCR recognizes self peptide-MHC complexes (positive selection), but the affinity for these complexes is not too high (negative selection), will the T cell mature and enter the blood stream [31]. Consequently, T cells specific for TAA or CTA express TCRs of weak TCR–peptid/MHC complex (pMHC) affinity. For example, while affinities, in terms of dissociation constants (KD), ranged between 0.18 and 25 μM (mean = 8.25 μM) for TCR from virus-specific CTLs, it was 10.7 to 387 μM (mean = 96.6 μM) for TAA-specific TCRs [32]. Experiments with T cells expressing a panel of affinity-optimized TCRs revealed that those T cells expressing TCRs with affinities lying in the upper physiological range (KD from 5 to 1 μM) demonstrated the best biological responses. When T cells expressed TCR with supraphysiological affinities (KD < 1 μM), their function decreased drastically. Hence, T cell activation and subsequent function benefit from enhanced TCR-pMHC affinities as long as they are in certain range (reviewed in [33]). Importantly, since neoantigen expression is restricted to tumor tissue, TCR affinities of neoantigen-specific T cells should span the entire physiological range including high-affinity TCRs often found for non-self specificities [33]. Thus, the presence of neoantigens might result in stronger antitumor immune responses. Support for this hypothesis came from a study of TCGA data. The authors calculated the immune cytotolytic activity of tumors based on transcript levels of granzyme A and perforin. For multiple tumor entities, they revealed a significant positive association of immune cytolytic activity with both mutation load and neoantigen prediction [34]. It should be noted, however, that for example, in melanoma T cell, reactivity against TAA is frequently observed and can also be broadened by adoptive T-cell transfer or anti-CTLA-4 treatment [35, 36].
TCR rearrangement and clonal expansion
T cell development takes place in the thymus (Fig. 1). T cell precursor cells lacking most of surface molecules characteristic for mature T cells like CD4, CD8, or TCR travel from the bone marrow into the thymus. These cells are referred to as double negative (DN) thymocytes since they do not express CD4 or CD8. They can be further subdivided into four stages of differentiation based on CD44 and CD25 expression in mice while in humans, a different and larger set of markers are required for this differentiation including CD34, CD38, CD1a, and CD4 [37, 38]. For αβ TCR T cells, the first somatic rearrangement of TCR genes occur at the CD44−CD25+ DN3 stage with recombining diversity (D)β to joining (J)β.gene segments followed by variable (V) β to D-J joining at the CD44−CD25− DN4 lymphocyte stage [39]. After transcription, RNA processing, translation, and further processing and glycosylation, the thymocytes express a pre-TCR (preTCR) if a functional TCRβ was generated. This preTCR consists of a TCRβ-chain and an invariant preTCRα-chain and is associated with CD3 molecules transducing activation molecules to the cell. Although a ligand-independent model of preTCR activation has been favored so far, it was recently demonstrated that the β-subunit can bind to pMHC and this interaction can lead to Ca2+ flux which is critical for TCR signaling [40]. Pre-TCR signaling terminates TCRβ rearrangements ensuring allelic exclusion, rescues cells from apoptosis, contributes to the largest proliferative expansion during T cell development, enables DN thymocytes to differentiate to double positive (DP) thymocytes expressing CD4 and CD8, and triggers TCRα rearrangements. Hence, preTCR which not only respond to ligand-independent signaling but also to preTCR–pMHC interactions might proliferate more rapidly, increasing the representation of these particular β-clonotypes. This enrichment of β-clonotypes that are more predisposed to bind pMHC should optimize the αβTCR repertoire before the DP thymocyte stage [41]. Soon after, the preTCR checkpoint also known as β-selection, the rearrangement of the TCRα chain, i.e., V-J joining, occurs. For the TCRα chain, allelic exclusion is much lesser strict. Analyses in mice demonstrated that in about 30% of αβ T cells biallelic in frame rearrangements of the TCRα chain can be detected while only in 2–10% for the TCRβ chain. Due to TCRα/TCRβ pairing constraints and regulated TCRα chain turnover, TCRβ chain translation and posttranslational processing, the frequency of cells with two TCRs is about 10% for those with two TCRα chains and 1–3% for those with two TCRβ chains [42]. Anyway, at the DP status, the thymocytes express the TCR and they undergo positive and negative selection. Notably, most of the thymocytes fail to pass β-selection as well as positive and negative selection resulting in the loss of the majority of thymocytes, i.e., up to 95% [43]. It is a matter of debate to which extend positive and negative selection contribute to this loss. Recently, data obtained from a mouse model revealed that about six times more cells undergo negative selection than complete positive selection demonstrating that the majority of developing T cells is self-reactive [44].

αβ-T cell development in the thymus. Bone marrow-derived lymphoid precursors enter the thymus. These early committed T cells are termed double negative (DN) since they do not express CD4 and CD8 and can be subdivided into four stages of differentiation. At the DN3 stage, the αβ-T cell development is initiated with the joining of diversity (D) and joining (J) gene segments for the TCRβ chain followed by joining of variable (V) gene segment. These cells express a pre-TCR (preTCR) consisting of a TCRβ-chain and an invariant preTCRα-chain if a functional TCRβ was generated. Lacking preTCR expression leads to apoptosis of the respective cells. The remaining cells proliferate and TCRα rearrangement takes place resulting in cells expressing TCRs, CD4, and CD8 and are termed double positive (DP). If no TCR is expressed, or the TCR does not recognize self MHC class molecules, the cells die by neglect (positive selection). Cortical thymic epithelial cells (cTEC) serve as antigen-presenting cell which due to a unique proteolytic pathway can present private peptides. Self-MHC-specific T cells migrate to the medulla where they as single positive (SP) cells encounter self-peptide presented by medullary thymic epithelial cells (mTECs) or dendritic cells (DCs). If their TCR has a too high affinity for self-peptide/MHC complex, the cells will die (negative selection). The remaining cells leave as naïve T cells the thymus
From the DP state, thymocytes develop into single positive (SP) cells which either express the coreceptor CD4 or CD8. Currently, the model is favored that by engaging the TCR on DP thymocytes, the expression of CD8 is decreased while CD4 expression is increased. When the TCR recognizes a peptid/MHC class II complex, the cell is additionally stimulated by the engagement of the CD4 coreceptor which results in a CD4 SP T cells. In the absence of enhanced stimulation, CD8 expression is induced and CD4 expression inhibited giving rise to a CD8 SP T cell [45]. Interestingly, TCR sequence might influence the lineage commitment; studies on naïve T cells demonstrated that most V and J genes are strongly biased in the subsets with some segments increasing the odds for a certain lineage fivefold [46].
In order to respond to a wide variety of pathogens, T cells have to be able to recognize many different non-self peptides. Thus, T cells should express a broad diversity of unique TCRs. This diversity is not only mediated by combining the different gene segments during TCR gene rearrangements, i.e., in humans, 40–48 functional TRBV, 2 TRBD, 12–13 TRBJ, and 2 TRBC genes for TCRβ chain and 44–46 TRAV, 50 TRAJ, and 1TRAC genes for TCRα chain (www.imgt.org), but also by the junctional diversity which results from addition of P(alindromic)- and N(on-template)-nucleotides and/or deletion of N-nucleotides at the junctions between V, J, and D genes. Theoretically, the potential diversity created by the recombination machinery was estimated to be up to 1015 [47]. In reality, this diversity is much lower. Indeed, Vanhanen and colleagues recently analyzed the TCR repertoire in human thymus. By applying massive parallel sequencing to samples from four pediatric donors, they revealed that approximately 80% of TCRβ and 32% of TCRα sequences were productive. Since the TCRs of only a fraction of cells could be analyzed, they used different algorithms to estimate the total thymic repertoire to consist of 40 to 70 × 106 unique TCRβ sequences and 60 to 100 × 106 TCRα sequences [48]. For one donor, they had both thymus and blood sample. Analyzing of these samples demonstrated that only 5% of peripheral TCRβ sequences were also detected in the 20% of the total thymus diversity sequenced, implying that the clonal composition in the thymus is very transient. Since it is technically not possible to determine the complete repertoire present in the periphery, the total number of TCR clonotypes present in an individual has to be estimated from the observed clonotypes. The TCR repertoire can be characterized by richness, i.e., number of unique TCR sequences, and clonality. In this regard, the minimal estimate for unique TCRβ sequences for both CD4+ and CD8+ naïve T cells in young adults was 100 × 106. With age, this naïve repertoire richness declined 2- to 5-fold, but elderly healthy individuals still seem to possess a diverse T cell repertoire of 8–57 × 106 unique sequences [49]. Naïve T cell circulate throughout the body and interact within lymphoid organs with antigen presenting cells (APCs). If they encounter an APC presenting the antigen for which they express the specific receptor, these cells get primed as long as additional costimulatory signals (surface molecules and cytokines) are provided (Fig. 2a). This activation of T cells is followed by clonal expansion of the antigen-specific cells and differentiation into effector and memory T cells. Accordingly, the richness of memory cells is lower than in naïve cells. For example, richness in CD4+ memory compartment has been reported to be 50-fold lower than in the respective naïve repertoire. In CD8+, memory cells richness is even 5–10-fold lower than that estimated for the CD4+ memory cells [49]. In contrast, clonal sizes of memory cells was larger than for naïve T cells with CD8+ exceeding CD4+ memory T cells in clonal size. This might also explain why stratification of T cells solely based on CD4 and CD8 expression returned an about five times higher richness for CD4+ cells [50]. This study also demonstrated that there was almost no overlap in TCRβ sequences between CD4+ and CD8+ T cells. Moreover, based on the CD4/CD8 distinction, there was a preferential use of distinct amino acids in the complementary-determining region 3 (CDR3), i.e., the main CDR responsible for recognizing processed antigen which is encoded by V–D–J for TCRβ or V–J for TCRα including junctional diversity. Similarly, it has been reported that the electrostatic charge of CDR3 is different between CD4 and CD8 positive cells, with a positively charged CDR3 associated with CD4+ lineage and a negatively charged CDR3 with CD8+ lineage [46].

T-cell clonal expansion and TCR clonotype analysis. a For naïve T cells to become activated, they have to get three signals from mature dendritic cells (DCs). First, the T cell has to recognize the antigen through its TCR. Second, the T cell has to receive a costimulatory signal from the DC. The most prominent costimulatore receptor on T cells is CD28 interacting with CD80 and CD86 on DC. Third, cytokines have to stimulate cytokine receptors. For the clonal expansion of T cells, this cytokine is IL-2. b Interaction of TCR with peptid/MHC class I receptor. Each chain contains three complementarity-determining regions (CDRs). The CDR3 region is the most variable region due to junctional diversity (N), i.e., the random insertion and deletion of nucleotides at the rearrangement positions. CDR1 and 2 interact with the MHC molecule while CDR3 encodes the receptor portion that makes the majority of TCR contacts with antigenic peptides bound by MHC. Since the CDR3 is unique for every T cell clone, most assays determining T cell clonality are based on analyzing the CDR3 of the β-chain. c Three methods to determine clonality within a T cell population. On the left side, clonotype mapping is depicted. This method is based on the amplification of each Vb family with a Vb-specific primer and a common C-specific primer extended by a GC clamp. PCR products are analyzed by denaturing gradient gel electrophoresis (DGGE). When reaching the melting temperature of the amplicon which is dependent on the length and sequence of the CDR3 region, DNA strands separate and form—due to the GC clamp—a y-structure which moves very slowly in the gel. Each T cell clonotype is indicated by a distinct band in the gel (in the example a monoclonal T-cell population). In the middle, spectratyping is depicted which is based on resolving the length of CDR3 region and on the right a massive parallel sequencing approach. For clonotype mapping, mRNA is used, while the two other can be performed on DNA or mRNA. For the latter, after cDNA generation either multiplex PCR can be performed similar as for gDNA or 5′ rapid amplification of cDNA ends (RACE)
TCR repertoire usage and cancer
Characterizing the interaction of cancer cells with the host adaptive immune system is critical for understanding tumor immunology particularly with respect to identifying therapeutic targets and monitoring immunotherapies. Based on the attainment and function of the TCR, the TCR repertoire is a mirror of the human immune responses during infections, autoimmunity, aging, and cancer. Consequently, the TCR repertoire usage affects the course of diseases and is likely to serve as a predictive marker for immunomodulatory therapies.
Early observations on the TCR repertoire usage in melanoma determined by reverse transcriptase polymerase chain reaction (RT-PCR) for the different TCR Vβ families, subsequent denaturing gradient gel electrophoresis (DGGE) separating the amplicons both by length and sequence of the CDR3 (clonotype mapping), and direct sequencing of RT-PCR products demonstrated that highly expressed TCR were present both in the regressive and the progressive regions of the primary tumor [51]. Moreover, TCR sequences representing clonally expanded T cells at the tumor site could also be detected in the lymph nodes draining the tumors even in the absence of tumor cells [52]. Alternative methods introduced at that time to establish the TCR repertoire usage were either measuring the CDR3 length directly (spectratyping) [53] or after one further round of PCR using primers for the J-region (Immunoscope) (Fig. 2c) [54].
However, characterizing the size and complexity of TCR repertoires has been a long-standing challenge in immunology. Only recently, advances in both multiplex PCR and massive parallel sequencing techniques allowed the development of new technologies for analyzing the TCR repertoire usage with more ease [55]. By means of these technologies, it was possible not only to establish the TCR repertoire usage for T cell infiltrating the tumor (tumor infiltrating lymphocytes, TIL) but also to track dominant TCR clonotypes in blood or other metastatic tumor lesions over time. For example, when Bai et al. scrutinized the TCR repertoires of pancreatic cancers in both primary tumors and matched blood samples, they identified two clusters of patients with respect to the diversity of TCR repertoires [56]. In addition, clonotypes with low frequencies were found in significantly higher numbers in tumors as compared to the respective blood samples. In a study in ovarian cancer patients, the TCR repertoire analysis was not only stratified by primary cancers, ascites, and blood but also by T cell subpopulations, i.e., CD4+, CD8+, and regulatory T cells [57]. An enrichment of certain T cells in tumors and ascites was evident, but these dominant TCR clonotypes were only rarely overlapping in the different locations. As would be expected, the TCR sequences in the different T cell subtypes were mutually exclusive among the three subgroups. Comparison of the TCR repertoires of tumor-infiltrating T cells in malignant brain tumors with that of peripheral blood revealed that TCR of tumor-infiltrating T cells not only possess greater VJ-independent diversity than TCR of peripheral blood but also diverge from these on the basis of this VJ-independent diversity [58].
TCR repertoire as a tool to monitor adaptive immune responses against solid tumors
Immunotherapeutic interventions enhancing adaptive immune responses against solid tumors are on the rise and show remarkable responses in a subset of patients [59]. Currently, the most promising approaches in this regard are the use of checkpoint blocking antibodies (anti PD-L1, anti-PD-1, and anti-CTLA-4) alone or in combination and adoptive T cell transfer of in vitro expanded TILs or engineered T cells with tumor antigen-specific TCRs or chimeric antigen receptors (CARs) [60]. Given the high costs, the restricted efficacies to a subset of patients and the immune-related adverse events associated with some of these therapies, much effort is invested to identify markers to predict response and to determine the effectiveness of the respective therapy. Consequently, robust monitoring strategies of adaptive immune responses have to be established. One possibility is to characterize the tumor infiltrate. In this regard, in 2006, it was established that the type, density, and location of T cells within colorectal tumors could predict clinical outcomes better than any previously used staging system [61]. This immunoscore to assess antitumor immune responses was determined from formalin-fixed paraffin-embedded (FFPE) tumor material that contained a tumor margin as well as a central tumor region T cell infiltration. It was based on staining serial slides for CD3 (T cells) and CD45RO (memory T cells) and was slightly improved over time [62]. In melanoma, the establishment of a reliably immunoscore emerged to be more complicated. No correlations between intratumoral or peritumoral CD3, CD8, CD20, CD163, or FoxP3 cells with clinical outcome and response to immunotherapy was evident [62]. Despite the massive advances in immunotherapy, immunoscores to monitor immune responses towards tumors have not changed much within the last 10 years but are at the verge of being reformed [63]. In this regard, immune profiling and monitoring of tumors and peripheral immune cells has to be extended massively in order to grasp the dynamics in antitumor immune responses. A next generation of immune monitoring taking tumor heterogeneity, tumor microenvironment, immunologic phenotype of the tumor cells, activation state of immune cells, and clonal expansion of T cells into account has to be established.
In order to determine T-cell diversity and clonal expansions in the peripheral blood and in tumors, TCR repertoire analyses based on the distinctively identifiable TCR CDR3 regions can be applied. TCR repertoire analyses could be used to monitor dynamics of T cell clonality and to track individual tumor reactive T cell clones. Notably, however, the role of the clonal composition of those TILs as well as the T cell receptor repertoire usage in the peripheral blood is still largely unknown.
For an immunomodulatory therapy to be effective, T cells have to exert their function within the tumor. Consequently, TCR repertoire analyses of TIL might allow monitoring of tumor-specific adaptive immune responses, but studies here are scarce [64]. It has been described that patients with a more “clonal” T cell repertoire of TILs are more likely to respond to immunotherapy. Tumeh et al. reported that TILs with a more restricted TCRβ chain usage reflecting a more clonal T-cell population at baseline and an increased clonality under therapy was associated with response to anti-PD-1 blockade [65]. Based on those findings, it can be assumed that in the responders, a tumor-specific T cell response is negatively regulated by the interaction of PD-1 with its ligand PD-L1. In a study of patients with metastatic urothelial cancer treated with an anti-PD-L1 antibody, clonality in the tumor and expansion of dominant tumor-resident TCR clones in the blood of the patients upon therapy were associated with response. At baseline, a low peripheral TCR clonality was associated with survival [66]. Expansion of tumor-infiltrating T-cell clones is, however, not restricted to active immunomodulatory therapies. When Cooper et al. characterized in sequential analyses the TCR repertoire of TIL in tumor biopsies obtained before BRAF inhibitor treatment and up to 14 days under treatment, they observed a strong increase in clonality. Notably, the majority of individual T cell clones detected under BRAF inhibitor treatment were new clones, i.e., not detectable prior to treatment. Similarly as for the anti-PD-1 therapy, existence of preexisting dominant T cell clones was associated with therapy response [67].
When analyzing the TCR repertoire of TILs, it should be kept in mind that tumor heterogeneity might not only be present within cells of a tumor, between primary tumors and metastasis, and T cell infiltration but also in the TCR repertoire usage. For example, TCR repertoire usage differs largely between tumors and bordering normal tissue [68, 69]. Moreover, the TCR repertoire detected in primary tumors can differ substantially from their metastasis [70, 71]. A significant TIL TCR repertoire heterogeneity has been described in renal cell carcinoma [72], whereas it appeared to be more homogeneous in ovarian cancer [73]. In esophageal squamous cell carcinoma there seem to be some more abundant, ubiquitous clones that are usually present in all tumor areas, but other clones were locally dominant only in some areas [69]. Taking those findings into account, care must be applied by selecting samples of tumor and metastasis for TIL TCR repertoire immunomonitoring and should be accompanied by histological work up.
TCR repertoire analyses of TIL can be performed either directly from tumor samples or by isolation and short-term culture of the TILs. The latter approach offers the advantages that T cell specificity and single-cell analyses can be performed. TCR analysis studies on ex vivo expanded TILs, however, can introduce also a bias. Indeed, TCR diversity of TIL is decreased after in vitro culture over time as the frequency of those TILs is increased which are able to expand in vitro [74]. Thus, if accompanied TCR specificity studies are planned, TIL TCR chain sequencing should be performed on freshly isolated or at least short-term cultured TILs in order to monitor the TIL TCR repertoire as unbiased as possible. Another possible solution, also in regard of studying TCR functionality, could be the generation of TCR libraries from freshly isolated TILs [64].
For TCR receptor monitoring, peripheral blood samples are preferable because they are easy to obtain. This leads to the question whether tumor-reactive TILs can be monitored in the peripheral blood of cancer patients. So far, the majority of reports stated that there is no or only a small overlap between the dominant TIL clones and the peripheral blood TCR repertoire usage. Besides the studies already mentioned in the previous section, Emerson et al. reported for example that in ovarian carcinoma, the TIL TCR repertoire was distinct from the circulating immune repertoire [73]. Similarly, Chen et al. reported that in esophageal squamous cell carcinoma, the largest TIL clones were only rarely detectable in the patients’ blood [69]. This fact is not surprising as the TCR repertoire in blood is a fast-changing dynamic process with a high turnover rate. Moreover, TCR repertoire analyses can only be performed on a fraction of the total cell population and even reveals only a portion of available T cell clonotypes present in a blood sample. Indeed, when two libraries of the same blood samples were analyzed, there was only a 25% overlap of obtained sequences. Moreover, only 13% of clonotypes were identical between two independent blood samples [75]. Notably, detection of tumor-reactive T cells in the blood might also be impeded, when they expand within the tumor in tertiary lymphoid structures [76]. With improved techniques, however, repeated blood draws might still be an option to monitor expansion or shrinking of a tumor reactive clone in the peripheral blood of cancer patients. In this regard, analysis of the peripheral T-cell repertoire will be especially useful to monitor T cell dynamics after adoptive T cell transfer of peripheral T cells, ex vivo expanded TILs, or after transfer of T cells with engineered tumor reactive TCRs. In fact, autologous transferred T cells could already be monitored in patients with B cell malignancies based on their TCRβ chain sequence [77], in patients after hematopoietic stem cell transplantation [78] and in a melanoma patient receiving adoptive T cell transfer therapy [79].
Several studies have been performed to determine the TCR repertoire in the periphery at baseline and correlate these with therapy efficacy. For example, a severe restricted TCR repertoire diversity in the peripheral blood at baseline was reported to be associated with a poor prognosis in breast cancer patients receiving chemotherapy [80]. For melanoma, however, conflicting results have been published. While Robert et al. did not detect any differences in the baseline TCR diversity between responders and non-responders to CTLA-4 blockade [81], Postow and colleges reported that high baseline TCR diversity was associated with clinical benefit after CTLA-4 blockade [82]. The diverse results may be due to the fact that different CTLA-4 checkpoint blocker, i.e., ipilimumab and tremelimumab, were used. Indeed, it was recently reported that patients with a low diversity score of T cells in peripheral blood did benefit from anti-PD-1 therapy, but not from anti-CTLA-4 therapy [83]. Thus, baseline TCR diversity may be a predictive biomarker for certain therapies and analyzing the TCR diversity might provide the rational for the choice of the most promising checkpoint blocker for the respective patient.
As mentioned previously, immunotherapeutic interventions can alter the TCR repertoire in the peripheral blood. In this regard, CTLA-4 blockade increases the diversity of the TCR repertoire in the peripheral blood of patients with melanoma [81]. This finding was confirmed for melanoma and prostate cancer [84]. Cha and colleges further reported a general loss of clonotypes after CTLA-4 blockade, but if pretherapy existing high-frequency T cell clones are maintained under therapy, this was associated with a longer survival. Moreover, TIL therapy leads to a significant broadening of the melanoma-reactive T cell repertoire in the peripheral blood of melanoma patients [35], and the administration of interleukin 7 in humans resulted in a marked broadening of the peripheral TCR repertoire by expansion of naïve T cell subsets [85]. Similarly, epigenetic compounds such as DNA demethylating agents that are currently tested in combination with classical immunotherapeutic approaches can broaden the peripheral TCR repertoire [86], and treatment of prostate cancer patients with the vaccine sipuleucel-T diversified the TCR repertoire in the blood and tumors of those patients, i.e., reduced diversity in the blood and increased it in the tumor [87]. These initial studies demonstrating that TCR monitoring can detect changes in TCR diversity associated with clinical outcome warrants studies with larger patient cohorts.
Methods for TCR repertoire analysis
As mentioned above, comprehensive TCR repertoire analysis was already introduced in the early nineties. However, techniques such as “clonotype mapping,” “spectratyping,” and “immunoscope” were only used by a limited number of laboratories. Indeed, a wider application of TCR repertoire analysis took place only after introduction of massive parallel sequencing of the TCR (TCR-seq) [88, 89]. TCR-seq involves the use of multiplexed PCR and massive parallel sequencing to generate large numbers of short DNA sequences covering key regions of the TCR coding sequence containing the CDR3 region (Fig. 2b).
Notably, TCR-seq is not conceptually different from spectratyping or clonotype mapping. However, since all CDR3 amplification products are directly used for preparation of the sequencing library and subsequent bulk sequencing, TCR-seq enables quantification of TCR sequence diversity at an unprecedented resolution and fidelity. A completely alternative approach is taking advantage of available high-throughput sequencing data generated not specifically for TCR repertoire analyses. To this end, Li et al. by using the TCGA cohort demonstrated the feasibility of using unselected RNA-seq data to characterize the TCR repertoire of tumor-infiltrating T cells [90]. Although this approach is currently still limited by low coverage and insufficient sample size, it revealed several interesting associations between the TCR repertoire and clinical and molecular features of the tumor: e.g., the diversity of T cell clonotypes positively associates with cancer somatic mutation load and the association of T cell diversity with expression of cancer/testis antigens identified new vaccine targets.
TCR-seq can be performed on both T-cell DNA or RNA, with DNA being more often used due to its abundance, ease of isolation, and long-term stability. Indeed, TCR-seq can be even applied to FFPE tissue samples. Since for each TCR chain there are two chromosomal loci per cell, the number of DNA template molecules in the PCR reaction corresponds to the number of T cells expressing the respective TCR. However, for absolute quantification of specific TCR sequences, it should be kept in mind that the V and J gene-specific primers are combined in a highly multiplexed PCR reaction in order to capture the entire repertoire. Thus, due to differences in annealing and amplification efficiencies of the respective primer pairs, read counts can be affected by PCR amplification biases. TCR-seq on T-cell RNA is likely to emphasize the representation of activated T cells because the TCR is among the highly transcribed genes in response to T cell activation [91]. This advantage, however, is also the drawback of using T cell RNA for analysis, i.e., variation in TCR expression levels among T cells means that the copy number of TCR template molecules is not really proportional to the number of cell of the respective clone.
In general, TCR-seq is commonly limited to one chain, in general the TCR β chain due to the before mentioned greater combinatorial and junctional diversity. Furthermore, because of allelic exclusion, αβ TCRs are more likely to contain only a single productive β chain variant, thus the number of distinct β chain sequences is more stringent associated with the number of T cell clonotypes present in a sample. From a biological perspective, it is important to remember that functional antigen-engaging TCRs are heterodimers, i.e., TCR αβ or TCR γδ. Hence, for functional analysis of TCRs, both subunits must be defined within one individual cell or cell clone. Most TCR-seq protocols begin with cell lysis eradicating α and β chain pairing specificity. Several approaches have been recently reported aiming at addressing this notion, i.e., cell-based emulsion RT-PCR, pairSEQ, Drop-seq, or single-cell TCRseq (scTCR-seq):
(i) A new type of PCR suppression technique was developed which allows amplifying a fused library with minimal noise for subsequent massive parallel paired-end sequencing. This cell-based emulsion RT-PCR results in the selective fusion of the native pairs of amplified TCR alpha and beta chain genes for complex samples, thus enabling identification of multiple native TCR chain pairs in a single analysis of a complex blood sample [92, 93].
(ii) pairSEQ may leverage the diversity of TCR sequences to accurately pair hundreds of thousands of TCR α and β sequences obtained from T cells from both blood and solid tissues in a single experiment [94, 95]. This strategy uses combinatorics, rather than physical isolation, to match the pairs based on the observation that rearranged TCR α and TCR β nucleotide sequences are nearly unique for each clonal population of T cells.
(iii) Drop-seq is based on adding a unique barcode to the mRNA from each cell prior to sequencing. Current Drop-seq technology enables capture, amplification, and barcoding of the entire mRNA transcriptome of individual cells before high-throughput sequencing. In general, this works well for mRNA sequences close to the poly-A tail. This limitation can be overcome by generating beads bearing multiple transcript-specific capture sequences. By means of a reversible chain blocking group, Hanson et al. were able to synthesize DNA barcoded beads bearing capture sequences for the constant domains of the T-cell receptor α and β chain mRNAs [96]. These beads had been used to capture and pair TCRα and TCRβ sequences from total T cell RNA before reverse transcription and PCR amplification.
(iv) Based on single-cell RNA-seq data, scTCRseq is a computational method to identify and count RNA read mapping to specific TCR V and C region genes, followed by multiple alignment of read mapping to the V and C regions with enough count to create consensus V and C gene sequence “contigs” across which gap-filling is performed [97]. This allows the identification of a single cell’s TCR CDR3 regions.
Given the complexity of both the biological input and digital output, TCR repertoire analysis requires specialized computational tools for both (i) data processing, in which raw sequence reads are filtered, assembled, and corrected for errors, and subsequently, (ii) data analysis. Biological or immunological interpretation of TCR-seq data can be challenging. Most analysis workflows allow sequence annotation and some combination of measurements for diversity and clonal expansion. For these tasks, multiple software solutions are available and it is beyond the scope of this review to describe and rate these tools. Indeed, we only mention solutions we are using in our group. tcR is an R package for the advanced analysis of TCR repertoires after primary CDR3 sequence extraction from the raw sequencing reads [98]. It includes diversity measures, shared T cell receptor sequence identification, gene usage statistic computation, and other widely used methods. A stable version can be directly installed from the Comprehensive R Archive Network (http://cran.r-project.org/mirrors.html). The source code and development version are available at http://imminfo.github.io/tcr/.
Limitations of TCR sequencing to monitor T cell responses
Currently, TCRβ chain sequencing allows monitoring presence, quantity, and clonal expansion of intratumoral and peripheral T cells. However, this information alone will most likely not be sufficient to fully characterize adaptive immune responses. There are several limitations that have to be overcome either by the usage of new techniques or by combining TCRβ chain sequencing results with other markers. For example, it has to be determined whether clonally expanded T cells indeed recognize tumor-specific antigens in the individual patient. For melanoma, it has been reported that less than 1% of TILs recognize shared melanoma antigens, the other 99% are of unknown specificity but might be virus, neoantigen, or tumor microenvironment-specific T cells or “bystanders” [35, 74]. Importantly, the TCR diversity of TIL in most cancers might be influenced by the extent of somatic mutations present in the tumor [90]. Besides monitoring, TCR sequence information can also be used to create tumor-reactive T cells. In this context, neoantigen-specific T cells might be superior compared to T cells specific for TAA. While the latter can be generated for different patients, neoantigen-specific T cells will be most often be personalized. Although TCRβ chain sequencing can function as a unique identifier for a specific T cell, it cannot be used alone to determine antigen specificity or tumor reactivity of a specific T cell. As discussed above, adaptations of the methods are needed to obtain information on both TCR chains. It is conceivable that in the future, sequence information on alpha and beta chain might allow predicting peptide specificity of the TCR; currently, it still has to be done experimentally. Interestingly, high throughput methods are currently developed to validate TCR affinity and to determine effectiveness of T cell responses. The in situ TCR affinity and sequence test (iTAST) measures single-cell 2D TCR affinity and sequence directly from primary CD8+ T cells [99], and barcode technology is used for high throughput epitope recognition profiling [100]. These methods might help to close the gap between TCR repertoire and T cell function. Regarding TCR-gene therapy, a rapid, personalized approach that targets the unique set of antigens presented by the autologous tumor without the need to identify their immunologic reactivity has recently been described [101].
It should be further noted that the molecular analysis of T cells neglects their phenotypic state. Only when combined with marker staining and sorting detailed information is available on the respective T-cell clonotypes. For example, a clonal expansion of a cytotoxic T cell is certainly more advantageous for the patient than of a regulatory T cell. Hence, besides TCR sequencing, the T cell infiltrate within a tumor should also be characterized phenotypically.
As discussed before, tumor heterogeneity and the fact that only a fraction of the clonotypes present in the blood can be analyzed are natural limitations which might be reduced by analyzing biological duplicates. Moreover, for comparison between peripheral and intratumoral repertoire sizes, normalization between the samples has to be performed. And finally, PCR amplification might introduce errors affecting clonality analysis. For accurate T cell clone detection, such errors have to be identified and corrected for [102].
Conclusion
With the advance of massive parallel sequencing, TCR repertoire analysis became not only easier, but the achievable read depth allows for detection of even low-frequency TCR sequences. In this regard, recent studies demonstrated that this approach is suitable to evaluate the diversity or clonality of T cells both in blood and tumor samples. Moreover, especially for tumor samples, T cell clonotypes can be tracked over time and in different tumors. Since it is a rather new technique, further improvements regarding the generation, processing, and analysis of data are likely. Nevertheless, some studies have already reported promising associations of baseline TCR repertoire or TCR repertoire changes under therapy with therapy responses. Certainly, larger studies are required to confirm TCR diversity as predictive or surrogate markers. Due to the easy access and non-invasiveness of blood samples, these would be the preferred samples to analyze. Hence, studies addressing the relation of TCR repertoires in tumors and periphery are encouraged. In this regard, encouraging results came from a recent study demonstrating that TCR repertoires of the circulating and tumor-infiltrating CD8+PD-1+ cells appeared similar [103]. Thus, distinct peripheral T-cell subsets might mirror the immune response in the tumor, and analyzing these subsets could give insight into the dynamics of the ongoing immune response.
References
Busch W (1868) Verhandlungen artzlicher gesellschaften. Berl. Klin Wochenschr 5:137–138
Coley WB (1893) The treatment of malignant tumors by repeated inoculations of erysipelas: with a report of ten original cases. Am J Med Sci 105:487–511
Ehrlich P (1909) Über den jetzigen stand der karzinomforschung. Ned Tijdschr Geneeskd 5:273–290
Burnet FM (1970) The concept of immunological surveillance. Prog Exp Tumor Res 13:1–27
Boyle J (2008) Molecular biology of the cell, 5th edition by B. Alberts, a. Johnson, J. Lewis, M. Raff, K. Roberts, and P. Walter. Biochem Mol Biol Educ 36:317–318. doi:10.1002/bmb.20192
Hermaszewski RA, Webster AD (1993) Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. Q J Med 86:31–42
Liu Q, Yan L, Xu C, Gu A, Zhao P, Jiang ZY (2014) Increased incidence of head and neck cancer in liver transplant recipients: a meta-analysis. BMC Cancer 14(776). doi:10.1186/1471-2407-14-776
Robbins HA, Pfeiffer RM, Shiels MS, Li J, Hall HI, Engels EA (2015) Excess cancers among HIV-infected people in the United States. J Natl Cancer Inst 107. doi:10.1093/jnci/dju503
Shang W, Huang L, Li L, Li X, Zeng R, Ge S, Xu G (2016) Cancer risk in patients receiving renal replacement therapy: a meta-analysis of cohort studies. Mol Clin Oncol 5:315–325. doi:10.3892/mco.2016.952
Bajaj NS, Watt C, Hadjiliadis D, Gillespie C, Haas AR, Pochettino A, Mendez J, Sterman DH, Schuchter LM, Christie JD, Lee JC, Ahya VN (2010) Donor transmission of malignant melanoma in a lung transplant recipient 32 years after curative resection. Transpl Int 23:e26–e31. doi:10.1111/j.1432-2277.2010.01090.x
Kim JK, Carmody IC, Cohen AJ, Loss GE (2009) Donor transmission of malignant melanoma to a liver graft recipient: case report and literature review. Clin Transpl 23:571–574. doi:10.1111/j.1399-0012.2008.00928.x
Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD (2007) Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450:903–907. doi:10.1038/nature06309
Giraldo NA, Becht E, Remark R, Damotte D, Sautes-Fridman C, Fridman WH (2014) The immune contexture of primary and metastatic human tumours. Curr Opin Immunol 27:8–15. doi:10.1016/j.coi.2014.01.001
Challis GB, Stam HJ (1990) The spontaneous regression of cancer. A review of cases from 1900 to 1987. Acta Oncol 29:545–550
Iannello A, Raulet DH (2013) Immune surveillance of unhealthy cells by natural killer cells. Cold Spring Harb Symp Quant Biol 78:249–257. doi:10.1101/sqb.2013.78.020255
Hayashi T, Imai K, Morishita Y, Hayashi I, Kusunoki Y, Nakachi K (2006) Identification of the NKG2D haplotypes associated with natural cytotoxic activity of peripheral blood lymphocytes and cancer immunosurveillance. Cancer Res 66:563–570. doi:10.1158/0008-5472.CAN-05-2776
Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, Knoblaugh S, Cado D, Greenberg NM, Raulet DH (2008) NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 28:571–580. doi:10.1016/j.immuni.2008.02.016
Pasero C, Gravis G, Guerin M, Granjeaud S, Thomassin-Piana J, Rocchi P, Paciencia-Gros M, Poizat F, Bentobji M, Azario-Cheillan F, Walz J, Salem N, Brunelle S, Moretta A, Olive D (2016) Inherent and tumor-driven immune tolerance in the prostate microenvironment impairs natural killer cell antitumor activity. Cancer Res 76:2153–2165. doi:10.1158/0008-5472.CAN-15-1965
Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, Koch M, Weitz J, Kloor M, Zoernig I, Schirmacher P, Brand K, Grabe N, Falk CS (2011) Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res 17:678–689. doi:10.1158/1078-0432.CCR-10-2173
Del Mar Valenzuela-Membrives M, Perea-Garcia F, Sanchez-Palencia A, Ruiz-Cabello F, Gomez-Morales M, Miranda-Leon MT, Galindo-Angel I, Farez-Vidal ME (2016) Progressive changes in composition of lymphocytes in lung tissues from patients with non-small-cell lung cancer. Oncotarget. doi:10.18632/oncotarget.12264
Pedersen L, Idorn M, Olofsson GH, Lauenborg B, Nookaew I, Hansen RH, Johannesen HH, Becker JC, Pedersen KS, Dethlefsen C, Nielsen J, Gehl J, Pedersen BK, Thor Straten P, Hojman P (2016) Voluntary running suppresses tumor growth through epinephrine- and IL-6-dependent NK cell mobilization and redistribution. Cell Metab 23:554–562. doi:10.1016/j.cmet.2016.01.011
Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF (2009) Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 69:3077–3085. doi:10.1158/0008-5472.CAN-08-2281
Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF (2011) Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha} + dendritic cells. J Exp Med 208:2005–2016. doi:10.1084/jem.20101159
Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre ML, Gajewski TF (2014) STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41:830–842. doi:10.1016/j.immuni.2014.10.017
Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD (2011) Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208:1989–2003. doi:10.1084/jem.20101158
Lurquin C, Van Pel A, Mariame B, De Plaen E, Szikora JP, Janssens C, Reddehase MJ, Lejeune J, Boon T (1989) Structure of the gene of tum- transplantation antigen P91A: the mutated exon encodes a peptide recognized with Ld by cytolytic T cells. Cell 58:293–303
van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, Boon T (1991) A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254:1643–1647
Trolle T, McMurtrey CP, Sidney J, Bardet W, Osborn SC, Kaever T, Sette A, Hildebrand WH, Nielsen M, Peters B (2016) The length distribution of class I-restricted T cell epitopes is determined by both peptide supply and MHC allele-specific binding preference. J Immunol 196:1480–1487. doi:10.4049/jimmunol.1501721
YC L, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, Davis L, Dudley ME, Yang JC, Samuels Y, Rosenberg SA, Robbins PF (2014) Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res 20:3401–3410. doi:10.1158/1078-0432.CCR-14-0433
Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348:69–74. doi:10.1126/science.aaa4971
Klein L, Kyewski B, Allen PM, Hogquist KA (2014) Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol 14:377–391. doi:10.1038/nri3667
Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM, Jakobsen BK (2012) Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol 42:3174–3179. doi:10.1002/eji.201242606
Hebeisen M, Allard M, Gannon PO, Schmidt J, Speiser DE, Rufer N (2015) Identifying individual T cell receptors of optimal avidity for tumor antigens. Front Immunol 6:582. doi:10.3389/fimmu.2015.00582
Rooney MS, Shukla SA, CJ W, Getz G, Hacohen N (2015) Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160:48–61. doi:10.1016/j.cell.2014.12.033
Kvistborg P, Shu CJ, Heemskerk B, Fankhauser M, Thrue CA, Toebes M, van Rooij N, Linnemann C, van Buuren MM, Urbanus JH, Beltman JB, Thor Straten P, Li YF, Robbins PF, Besser MJ, Schachter J, Kenter GG, Dudley ME, Rosenberg SA, Haanen JB, Hadrup SR, Schumacher TN (2012) TIL therapy broadens the tumor-reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunology 1:409–418
Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, Welters MJ, van der Burg S, Kapiteijn E, Michielin O, Romano E, Linnemann C, Speiser D, Blank C, Haanen JB, Schumacher TN (2014) Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med 6:254ra128. doi:10.1126/scitranslmed.3008918
Godfrey DI, Kennedy J, Suda T, Zlotnik A (1993) A developmental pathway involving four phenotypically and functionally distinct subsets of CD3-CD4-CD8- triple-negative adult mouse thymocytes defined by CD44 and CD25 expression. J Immunol 150:4244–4252
Staal FJ, Weerkamp F, Langerak AW, Hendriks RW, Clevers HC (2001) Transcriptional control of t lymphocyte differentiation. Stem Cells 19:165–179. doi:10.1634/stemcells.19-3-165
Livak F, Tourigny M, Schatz DG, Petrie HT (1999) Characterization of TCR gene rearrangements during adult murine T cell development. J Immunol 162:2575–2580
Mallis RJ, Bai K, Arthanari H, Hussey RE, Handley M, Li Z, Chingozha L, Duke-Cohan JS, Lu H, Wang JH, Zhu C, Wagner G, Reinherz EL (2015) Pre-TCR ligand binding impacts thymocyte development before alphabetaTCR expression. Proc Natl Acad Sci U S A 112:8373–8378. doi:10.1073/pnas.1504971112
Yang X, Mariuzza RA (2015) Pre-T-cell receptor binds MHC: implications for thymocyte signaling and selection. Proc Natl Acad Sci U S A 112:8166–8167. doi:10.1073/pnas.1510127112
Brady BL, Steinel NC, Bassing CH (2010) Antigen receptor allelic exclusion: an update and reappraisal. J Immunol 185:3801–3808. doi:10.4049/jimmunol.1001158
Kyewski B, Klein L (2006) A central role for central tolerance. Annu Rev Immunol 24:571–606. doi:10.1146/annurev.immunol.23.021704.115601
Stritesky GL, Xing Y, Erickson JR, Kalekar LA, Wang X, Mueller DL, Jameson SC, Hogquist KA (2013) Murine thymic selection quantified using a unique method to capture deleted T cells. Proc Natl Acad Sci U S A 110:4679–4684. doi:10.1073/pnas.1217532110
Singer A, Adoro S, Park JH (2008) Lineage fate and intense debate: myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat Rev Immunol 8:788–801. doi:10.1038/nri2416
Klarenbeek PL, Doorenspleet ME, Esveldt RE, van Schaik BD, Lardy N, van Kampen AH, Tak PP, Plenge RM, Baas F, de Bakker PI, de Vries N (2015) Somatic variation of T-cell receptor genes strongly associate with HLA class restriction. PLoS One 10:e0140815. doi:10.1371/journal.pone.0140815
Davis MM, Bjorkman PJ (1988) T-cell antigen receptor genes and T-cell recognition. Nature 334:395–402. doi:10.1038/334395a0
Vanhanen R, Heikkila N, Aggarwal K, Hamm D, Tarkkila H, Patila T, Jokiranta TS, Saramaki J, Arstila TP (2016) T cell receptor diversity in the human thymus. Mol Immunol 76:116–122. doi:10.1016/j.molimm.2016.07.002
Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee JY, Olshen RA, Weyand CM, Boyd SD, Goronzy JJ (2014) Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci U S A 111:13139–13144. doi:10.1073/pnas.1409155111
Li HM, Hiroi T, Zhang Y, Shi A, Chen G, De S, Metter EJ, Wood WH 3rd, Sharov A, Milner JD, Becker KG, Zhan M, Weng NP (2016) TCRbeta repertoire of CD4+ and CD8+ T cells is distinct in richness, distribution, and CDR3 amino acid composition. J Leukoc Biol 99:505–513. doi:10.1189/jlb.6A0215-071RR
thor Straten P, Becker JC, Seremet T, Brocker EB, Zeuthen J (1996) Clonal T cell responses in tumor infiltrating lymphocytes from both regressive and progressive regions of primary human malignant melanoma. J Clin Invest 98:279–284. doi:10.1172/JCI118790
Straten P, Dahl C, Schrama D, Pedersen LO, Andersen MH, Seremet T, Brocker EB, Guldberg P, Becker JC (2006) Identification of identical TCRs in primary melanoma lesions and tumor free corresponding sentinel lymph nodes. Cancer Immunol Immunother 55:495–502. doi:10.1007/s00262-005-0023-8
Cochet M, Pannetier C, Regnault A, Darche S, Leclerc C, Kourilsky P (1992) Molecular detection and in vivo analysis of the specific T cell response to a protein antigen. Eur J Immunol 22:2639–2647. doi:10.1002/eji.1830221025
Pannetier C, Even J, Kourilsky P (1995) T-cell repertoire diversity and clonal expansions in normal and clinical samples. Immunol Today 16:176–181
Robins H (2013) Immunosequencing: applications of immune repertoire deep sequencing. Curr Opin Immunol 25:646–652. doi:10.1016/j.coi.2013.09.017
Bai X, Zhang Q, Wu S, Zhang X, Wang M, He F, Wei T, Yang J, Lou Y, Cai Z, Liang T (2015) Characteristics of tumor infiltrating lymphocyte and circulating lymphocyte repertoires in pancreatic cancer by the sequencing of T cell receptors. Sci Rep 5:13664. doi:10.1038/srep13664
Jang M, Yew PY, Hasegawa K, Ikeda Y, Fujiwara K, Fleming GF, Nakamura Y, Park JH (2015) Characterization of T cell repertoire of blood, tumor, and ascites in ovarian cancer patients using next generation sequencing. Oncoimmunology 4:e1030561. doi:10.1080/2162402X.2015.1030561
Sims JS, Grinshpun B, Feng Y, Ung TH, Neira JA, Samanamud JL, Canoll P, Shen Y, Sims PA, Bruce JN (2016) Diversity and divergence of the glioma-infiltrating T-cell receptor repertoire. Proc Natl Acad Sci U S A 113:E3529–E3537. doi:10.1073/pnas.1601012113
Topalian SL, Wolchok JD, Chan TA, Mellman I, Palucka K, Banchereau J, Rosenberg SA, Dane Wittrup K (2015) Immunotherapy: the path to win the war on cancer? Cell 161:185–186
Menon S, Shin S, Dy G (2016) Advances in cancer immunotherapy in solid tumors. Cancers 8:106. doi:10.3390/cancers8120106
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoué F, Bruneval P, Cugnenc P-H, Trajanoski Z, Fridman W-H, Pagès F (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (New York, NY) 313:1960–1964. doi:10.1126/science.1129139
Galon J, Fox BA, Bifulco CB, Masucci G, Rau T, Botti G, Marincola FM, Ciliberto G, Pages F, Ascierto PA, Capone M (2016) Immunoscore and immunoprofiling in cancer: an update from the melanoma and immunotherapy bridge 2015. J Transl Med 14:273. doi:10.1186/s12967-016-1029-z
Kohrt HE, Tumeh PC, Benson D, Bhardwaj N, Brody J, Formenti S, Fox BA, Galon J, June CH, Kalos M, Kirsch I, Kleen T, Kroemer G, Lanier L, Levy R, Lyerly HK, Maecker H, Marabelle A, Melenhorst J, Miller J, Melero I, Odunsi K, Palucka K, Peoples G, Ribas A, Robins H, Robinson W, Serafini T, Sondel P, Vivier E, Weber J, Wolchok J, Zitvogel L, Disis ML, Cheever MA, (CITN) CITN (2016) Immunodynamics: a cancer immunotherapy trials network review of immune monitoring in immuno-oncology clinical trials. Journal for immunotherapy of cancer 4:711. doi:10.1186/s40425-016-0118-0
Linnemann C, Mezzadra R, Schumacher TNM (2014) TCR repertoires of intratumoral T-cell subsets. Immunol Rev 257:72–82. doi:10.1111/imr.12140
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568–571. doi:10.1038/nature13954
Funt S, Charen AS, Yusko E, Vignali M, Benzeno S, Boyd ME, Moran MM, Kania BE, Cipolla CK, Regazzi AM, Robins H, Iyer G, Rosenberg JE, and Bajorin DF (2016) Correlation of peripheral and intratumoral T-cell receptor (TCR) clonality with clinical outcomes in patients with metastatic urothelial cancer (mUC) treated with atezolizumab. in ASCO Abstract 3005. J Clin Oncol
Cooper ZA, Frederick DT, Juneja VR, Sullivan RJ, Lawrence DP, Piris A, Sharpe AH, Fisher DE, Flaherty KT, Wargo JA (2013) BRAF inhibition is associated with increased clonality in tumor-infiltrating lymphocytes. Oncoimmunology 2:e26615. doi:10.4161/onci.26615
Sherwood AM, Emerson RO, Scherer D, Habermann N, Buck K, Staffa J, Desmarais C, Halama N, Jaeger D, Schirmacher P, Herpel E, Kloor M, Ulrich A, Schneider M, Ulrich CM, Robins H (2013) Tumor-infiltrating lymphocytes in colorectal tumors display a diversity of T cell receptor sequences that differ from the T cells in adjacent mucosal tissue. Cancer immunology, immunotherapy : CII 62:1453–1461. doi:10.1007/s00262-013-1446-2
Chen Z, Zhang C, Pan Y, Xu R, Xu C, Chen Z, Lu Z, Ke Y (2016) T cell receptor β-chain repertoire analysis reveals intratumour heterogeneity of tumour-infiltrating lymphocytes in oesophageal squamous cell carcinoma. J Pathol 239:450–458. doi:10.1002/path.4742
Willhauck M, Möhler T, Scheibenbogen C, Pawlita M, Brossart P, Schmier JW, Keilholz U (1996) T-cell receptor beta variable region diversity in melanoma metastases after interleukin 2-based immunotherapy. Clinical cancer research : an official journal of the American Association for Cancer Research 2:767–772
Hishii M, Andrews D, Boyle LA, Wong JT, Pandolfi F, van den Elsen PJ, Kurnick JT (1997) In vivo accumulation of the same anti-melanoma T cell clone in two different metastatic sites. Proc Natl Acad Sci 94:1378–1383
Gerlinger M, Quezada SA, Peggs KS, Furness AJS, Fisher R, Marafioti T, Shende VH, McGranahan N, Rowan AJ, Hazell S, Hamm D, Robins HS, Pickering L, Gore M, Nicol DL, Larkin J, Swanton C (2013) Ultra-deep T cell receptor sequencing reveals the complexity and intratumour heterogeneity of T cell clones in renal cell carcinomas. J Pathol 231:424–432. doi:10.1002/path.4284
Emerson RO, Sherwood AM, Rieder MJ, Guenthoer J, Williamson DW, Carlson CS, Drescher CW, Tewari M, Bielas JH, Robins HS (2013) High-throughput sequencing of T-cell receptors reveals a homogeneous repertoire of tumour-infiltrating lymphocytes in ovarian cancer. J Pathol 231:433–440. doi:10.1002/path.4260
Andersen RS, Thrue CA, Junker N, Lyngaa R, Donia M, Ellebæk E, Svane IM, Schumacher TN, Thor Straten P, Hadrup SR (2012) Dissection of T-cell antigen specificity in human melanoma. Cancer Res 72:1642–1650. doi:10.1158/0008-5472.CAN-11-2614
Warren RL, Freeman JD, Zeng T, Choe G, Munro S, Moore R, Webb JR, Holt RA (2011) Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res 21:790–797. doi:10.1101/gr.115428.110
Dieu-Nosjean M-C, Goc J, Giraldo NA, Sautès-Fridman C, Fridman W-H (2014) Tertiary lymphoid structures in cancer and beyond. Trends Immunol 35:571–580. doi:10.1016/j.it.2014.09.006
Grupp SA, Prak EL, Boyer J, McDonald KR, Shusterman S, Thompson E, Callahan C, Jawad AF, Levine BL, June CH, Sullivan KE (2012) Adoptive transfer of autologous T cells improves T-cell repertoire diversity and long-term B-cell function in pediatric patients with neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research 18:6732–6741. doi:10.1158/1078-0432.CCR-12-1432
Li Y, Xu L (2015) Evaluation of TCR repertoire diversity in patients after hematopoietic stem cell transplantation. Stem cell investigation 2:17. doi:10.3978/j.issn.2306-9759.2015.09.01
Zhou J, Dudley ME, Rosenberg SA, Robbins PF (2005) Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. Journal of immunotherapy (Hagerstown, Md : 1997) 28:53–62
Manuel M, Tredan O, Bachelot T, Clapisson G, Courtier A, Parmentier G, Rabeony T, Grives A, Perez S, Mouret J-F, Perol D, Chabaud S, Ray-Coquard I, Labidi-Galy I, Heudel P, Pierga J-Y, Caux C, Blay J-Y, Pasqual N, Ménétrier-Caux C (2012) Lymphopenia combined with low TCR diversity (divpenia) predicts poor overall survival in metastatic breast cancer patients. OncoImmunology 1:432–440
Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, Mok S, Huang RR, Cochran AJ, Comin-Anduix B, Koya RC, Graeber TG, Robins H, Ribas A (2014) CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clinical cancer research : an official journal of the American Association for Cancer Research 20:2424–2432. doi:10.1158/1078-0432.CCR-13-2648
Postow MA, Manuel M, Wong P, Yuan J, Dong Z, Liu C, Perez S, Tanneau I, Noel M, Courtier A, Pasqual N, Wolchok JD (2015) Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. Journal for immunotherapy of cancer 3:23. doi:10.1186/s40425-015-0070-4
Goldinger SM, Courtier A, Jaberg-Bentele NF, Schindler S, Manuel M, Plantier N, Treillard B, Noel M, Nguyen-Kim TDL, Raaijmakers MIG, Kvistborg P, Haanen JBAG, Dummer R, and Levesque M (2016) The peripheral blood TCR repertoire to facilitate patient stratification for immune checkpoint blockade inhibition in metastatic melanoma. in ASCO Abstract 3026. J Clin Oncol
Cha E, Klinger M, Hou Y, Cummings C, Ribas A, Faham M, Fong L (2014) Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci Transl Med 6:238ra70. doi:10.1126/scitranslmed.3008211
Sportès C, Hakim FT, Memon SA, Zhang H, Chua KS, Brown MR, Fleisher TA, Krumlauf MC, Babb RR, Chow CK, Fry TJ, Engels J, Buffet R, Morre M, Amato RJ, Venzon DJ, Korngold R, Pecora A, Gress RE, Mackall CL (2008) Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J Exp Med 205:1701–1714. doi:10.1084/jem.20071681
Nie J, Zhang Y, Li X, Chen M, Liu C, Han W (2016) DNA demethylating agent decitabine broadens the peripheral T cell receptor repertoire. Oncotarget 7:37882–37892. doi:10.18632/oncotarget.9352
Sheikh N, Cham J, Zhang L, DeVries T, Letarte S, Pufnock J, Hamm D, Trager J, Fong L (2016) Clonotypic diversification of intratumoral T cells following sipuleucel-T treatment in prostate cancer subjects. Cancer Res 76:3711–3718. doi:10.1158/0008-5472.CAN-15-3173
Klinger M, Kong K, Moorhead M, Weng L, Zheng J, Faham M (2013) Combining next-generation sequencing and immune assays: a novel method for identification of antigen-specific T cells. PLoS One 8:e74231. doi:10.1371/journal.pone.0074231
Li S, Lefranc MP, Miles JJ, Alamyar E, Giudicelli V, Duroux P, Freeman JD, Corbin VD, Scheerlinck JP, Frohman MA, Cameron PU, Plebanski M, Loveland B, Burrows SR, Papenfuss AT, Gowans EJ (2013) IMGT/HighV QUEST paradigm for T cell receptor IMGT clonotype diversity and next generation repertoire immunoprofiling. Nat Commun 4:2333. doi:10.1038/ncomms3333
Li B, Li T, Pignon JC, Wang B, Wang J, Shukla SA, Dou R, Chen Q, Hodi FS, Choueiri TK, Wu C, Hacohen N, Signoretti S, Liu JS, Liu XS (2016) Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat Genet 48:725–732. doi:10.1038/ng.3581
Favier B, Burroughs NJ, Wedderburn L, Valitutti S (2001) TCR dynamics on the surface of living T cells. Int Immunol 13:1525–1532
Turchaninova MA, Britanova OV, Bolotin DA, Shugay M, Putintseva EV, Staroverov DB, Sharonov G, Shcherbo D, Zvyagin IV, Mamedov IZ, Linnemann C, Schumacher TN, Chudakov DM (2013) Pairing of T-cell receptor chains via emulsion PCR. Eur J Immunol 43:2507–2515. doi:10.1002/eji.201343453
Munson DJ, Egelston CA, Chiotti KE, Parra ZE, Bruno TC, Moore BL, Nakano TA, Simons DL, Jimenez G, Yim JH, Rozanov DV, Falta MT, Fontenot AP, Reynolds PR, Leach SM, Borges VF, Kappler JW, Spellman PT, Lee PP, Slansky JE (2016) Identification of shared TCR sequences from T cells in human breast cancer using emulsion RT-PCR. Proc Natl Acad Sci U S A 113:8272–8277. doi:10.1073/pnas.1606994113
Howie B, Sherwood AM, Berkebile AD, Berka J, Emerson RO, Williamson DW, Kirsch I, Vignali M, Rieder MJ, Carlson CS, Robins HS (2015) High-throughput pairing of T cell receptor alpha and beta sequences. Sci Transl Med 7:301ra131. doi:10.1126/scitranslmed.aac5624
Klinger M, Pepin F, Wilkins J, Asbury T, Wittkop T, Zheng J, Moorhead M, Faham M (2015) Multiplex identification of antigen-specific T cell receptors using a combination of immune assays and immune receptor sequencing. PLoS One 10:e0141561. doi:10.1371/journal.pone.0141561
Hanson WM, Chen Z, Jackson LK, Attaf M, Sewell AK, Heemstra JM, Phillips JD (2016) Reversible oligonucleotide chain blocking enables bead capture and amplification of T-cell receptor alpha and beta chain mRNAs. J Am Chem Soc 138:11073–11076. doi:10.1021/jacs.6b04465
Redmond D, Poran A, Elemento O (2016) Single-cell TCRseq: paired recovery of entire T-cell alpha and beta chain transcripts in T-cell receptors from single-cell RNAseq. Genome Med 8:80. doi:10.1186/s13073-016-0335-7
Nazarov VI, Pogorelyy MV, Komech EA, Zvyagin IV, Bolotin DA, Shugay M, Chudakov DM, Lebedev YB, Mamedov IZ (2015) tcR: an R package for T cell receptor repertoire advanced data analysis. BMC Bioinformatics 16:175. doi:10.1186/s12859-015-0613-1
Zhang S-Q, Parker P, Ma K-Y, He C, Shi Q, Cui Z, Williams CM, Wendel BS, Meriwether AI, Salazar MA, Jiang N (2016) Direct measurement of T cell receptor affinity and sequence from naïve antiviral T cells. Sci Transl Med 8:341ra77. doi:10.1126/scitranslmed.aaf1278
Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, Such L, Furness AJ, McGranahan N, Rosenthal R, Straten PT, Szallasi Z, Svane IM, Swanton C, Quezada SA, Jakobsen SN, Eklund AC, Hadrup SR (2016) Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat Biotechnol. doi:10.1038/nbt.3662
Pasetto A, Alena G, Robbins PF, Deniger DC, Prickett TD, Matus-Nicodemos R, Douek DC, Howie B, Robins H, Parkhurst MR, Gartner JJ, Trebska-McGowan K, Crystal JS, Rosenberg SA (2016) Tumor- and neoantigen-reactive T-cell receptors can be identified based on their frequency in fresh tumor. Cancer Immunol Res DOI. doi:10.1158/2326-6066.CIR-16-0001
Poschke I, Flossdorf M, Offringa R (2016) Next generation TCR sequencing—a tool to understand T cell infiltration in human cancers. J Pathol. doi:10.1002/path.4800
Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM, Trebska-McGowan K, Wunderlich JR, Yang JC, Rosenberg SA (2016) Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med 22:433–438. doi:10.1038/nm.4051
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Cancer and Autoimmunity - Guest Editor: Mads Hald Andersen
Rights and permissions
About this article

Cite this article
Schrama, D., Ritter, C. & Becker, J.C. T cell receptor repertoire usage in cancer as a surrogate marker for immune responses. Semin Immunopathol 39, 255–268 (2017). https://doi.org/10.1007/s00281-016-0614-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-016-0614-9