
Abstract
Dendritic cells are of paramount importance bridging innate and adaptive immune responses. Depending on the context, after sensing environmental antigens, commensal microorganisms, pathogenic agents, or antigens from the diet, dendritic cells may drive either different effector adaptive immune responses or tolerance, avoiding tissue damage. Although the plasticity of the immune response and the capacity to regulate itself are considered essential to orchestrate appropriate physiological responses, it is known that the nervous system plays a relevant role controlling immune cell function. Dendritic cells present in the skin, the intestine, and lymphoid organs, besides expressing adrenergic receptors, can be reached by neurotransmitters released by sympathetic fibers innervating these tissues. These review focus on how neurotransmitters from the sympathetic nervous system can modulate dendritic cell function and how this may impact the immune response and immune-mediated disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The immune system plays a crucial role maintaining general homeostasis and allowing physiological responses to occur in an appropriate manner. That includes a fine-tuned action distinguishing antigens that must be tolerated (such as those from the diet and commensal microbiota) from antigens that should elicit the generation of effector responses (such as those from pathogenic microorganisms). In addition, an effector response generated by the immune system will be physiologically appropriate as long as its magnitude and quality are adequate for each type of challenge or infection being faced by the host. Although the ability of the immune system to regulate itself is an important feature that contributes to the generation of appropriate physiological responses, the cross talk that takes place between the immune and the nervous systems has also been found to be very important in this sense. These two systems communicate in a bidirectional manner through the release of chemical messengers, such as cytokines and neurotransmitters [1, 2]. Exist, so far, three main described pathways by which the central nervous system (CNS) can influence the immune response: the hypothalamic–pituitary–adrenal (HPA) axis through glucocorticoid release [3], the sympathetic nervous system (SNS) that innervates all lymphoid organs and release neurotransmitters such as norepinephrine and dopamine [4], and through the vagal inflammatory reflex. Even though the spleen and lymph nodes do not receive innervation from parasympathetic fibers, the vagus nerve can modulate the immune response through a well-organized circuit, called inflammatory reflex, in which signals from the vagus nerve are thought to modulate the activity of the splenic nerve, which secretes norepinephrine within the spleen. Then, norepinephrine signaling via β2 adrenergic receptor (β2AR) on T cells induces the production and release of acetylcholine by these cells, which finally binds to α7nAChR expressed on splenic and other macrophages to effectively inhibit inflammatory cytokine release [5, 6]. The sympathetic nervous system, via norepinephrine release, can modulate several immunological processes, including lymphocyte ontogeny into the thymus, chemotaxis, antigen presentation and processing, and T cell function. In this review, we focus on how the sympathetic nervous system can impact aspects of the immune response, particularly those related to dendritic cell function and the consequences to the generation of adaptive immune responses and the development of immune-mediated diseases.
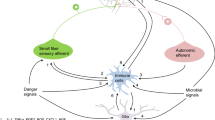
Dendritic cells and their interactions with sympathetic fibers in lymphoid organs, skin, and intestine
Dendritic cells (DCs) are potent antigen-presenting cells (APC) and play an important role in linking innate and adaptive immune responses as well as driving the immune response to immunity and tolerance. DCs can be classified into different populations according to their origin, location, activation status, and function. Thereby, based on their morphological and functional features, DCs have been classified in two major types: conventional or classical DCs (cDCs) and plasmacytoid DCs (pDCs) [7, 8]. Both cDCs and pDCs are derived from bone marrow progenitors in a Flt3L-dependent manner; however, each subset of DCs requires distinct transcription factors for its differentiation [9–11]. The cDCs are highly phagocytic cells, express high levels of major histocompatibility complex (MHC) class II molecules, and have potent antigen-presenting abilities, being important in the activation of naïve T cells. So far, cDCs have been divided in at least two major subsets characterized either by the expression of CD8α and CD103, or CD11b. CD8α+ cDCs from the spleen are specialized in cross-presenting exogenous antigens to CD8+ T cells, while CD11b+ cDCs preferentially activate CD4+ T cells [7, 8]. Both subsets are present in classical lymphoid organs, such as the spleen and lymph nodes, as well as in many other tissues, such as the skin and the gut [7, 8]. In many of the tissues that are not classically considered as lymphoid organs, the subset characterized as CD103+ does not express the CD8α molecule. Besides depending on Flt3L, the development of CD8α+ cDCs present in lymphoid organs and their equivalent populating other tissues, the CD103+ (CD11b−) cDCs, also relies on a transcription factor named basic leucine zipper ATF-like 3 (BATF3).
The pDCs represent a rare DC subset specialized in the production of type I interferon (IFN) in inflammatory conditions following toll-like receptor 7 (TLR7) or TLR9 activation. This subset of DCs presents lower capacity to perform phagocytosis and is considered inefficient inducers of CD4+ T cell responses [7, 8].
The DCs are responsible to recognize, process, and present antigens to T cells, tasks that often result in the activation and differentiation of T cells. The production and release of a distinct set of cytokines by DCs upon different types of stimulation is a central feature in determining the generation of diverse effector adaptive immune responses. In general, intracellular pathogens such as viruses and bacteria induce IL-12p70 production by DCs, which is linked with Th1 cell differentiation [12, 13]. In contrast, extracellular bacteria and fungi infections result in a production of high levels of IL-6 and IL-23, which can instead facilitate the orchestration of Th17-mediated immune responses [12, 14]. Depending on the context of antigen presentation, DCs are also important for tolerance induction and the differentiation of regulatory T cells [15].
Several studies were performed to characterize the sympathetic innervation of lymphoid organs such as the thymus, spleen, and lymph nodes.
Some of these studies were carried out using retrograde tracers, which are injected into the organ to be studied. The tracer is then retrogradely transported toward the cell body of neurons innervating such organ.
There were found positive stained neurons in sympathetic ganglia chain following injections made in the thymus [16, 17], spleen [18], and lymph nodes [19]. However, injections of such a tracer into the thymus or the spleen did not result in labeled neurons within the dorsal root ganglia of the spinal cord, indicating absence of afferent innervation in these organs [20]. Conversely, tracer injection in lymph nodes showed stained neurons in the dorsal root ganglia, suggesting afferent innervation in at least some lymph nodes [21].
Some other studies used an immunohistochemical approach to track for the presence of sympathetic and parasympathetic fibers. Usually, these techniques seek for the verification of immunoreactivity for transporters of neurotransmitters present in synaptic vesicles and enzymes associated with the synthesis of neurotransmitters. Among these enzymes are tyrosine hydroxylase (TH) and dopa β-hydroxylase (DBH), which are involved in noradrenaline and dopamine synthesis and therefore are present in sympathetic fibers, and choline acetyltransferase (ChAT), which participates in the synthesis of acetylcholine and is expressed in cholinergic fibers of the parasympathetic nervous system [20].
In fact, such lymphoid organs showed the presence of TH-positive and DBH-positive fibers, indicating the existence of catecholaminergic fibers [22]. Furthermore, the lack of positive fibers for vesicular acetylcholine transporters [23] and ChAT [24] in the thymus and the spleen indicates the absence of cholinergic fibers in such organs.
Altogether, these studies demonstrated a strong sympathetic innervation in the thymus, spleen, and lymph nodes and some sensory afferents in lymph nodes [reviewed in [20]].
Noradrenergic postganglionic innervation of the spleen originates in the superior mesenteric/celiac ganglion, and the fibers enter the spleen together with the blood vessels at the hilar region [18, 25]. The noradrenergic nerve fibers are distributed along the vascular and trabecular systems and travel mainly with the central artery and its branches, also extending prolongations into the parenchyma of the periarteriolar lymphatic sheath (PALS). Therefore, the white pulp, a region rich in T cell, is profusely innervated by sympathetic fibers. Electronic microscopy studies of TH+ fibers indicate close contact among lymphocytes, myeloid cells, and the terminal endings of nerve fibers [22].
Moreover, it was demonstrated by confocal microscopy that sympathetic nerve fiber endings are co-localized with macrophages in the spleen [26]. Since dendritic cells are also found within the white pulp of the spleen [27], in the same region where the T cells are, they are likely to be in tight contact with sympathetic nerve fibers. Confocal technology allied to immunohistochemical techniques also allowed the description of rare but very close interactions between follicular dendritic cell (FDC) network and sympathetic fibers within the spleen. The neural fibers were estimated to be closer than 10 μm from a FDC network [28].
In lymph nodes, sympathetic fibers are found in the medullary cords and the paracortical region, while they are quite sparse within follicles and close to germinal centers [22]. Thus, lymph nodes and the spleen receive sympathetic innervation in areas that are rich in T cells and dendritic cells, suggesting that events such as antigen processing, antigen presentation, and cell trafficking may be directly influenced by NA released from sympathetic fibers [22].
The skin is a morphologically complex organ with several complementary functions, including being a protective barrier, carrying a variety of mechanical, thermal, chemical, and noxious sensors, and playing an important role in thermoregulation, which is mediated by a rich vasculature that is innervated by sympathetic and sensory fiber endings [29]. Sympathetic innervation extends through dermis and epidermis [30, 31].
The skin also bears a huge collection of immune cells that are present in the epidermis and dermis, being important to detect and respond to tissue injury and infections. Moreover, because skin is the second largest surface area functioning as a barrier to the outside environment, the local immune system has to deal with a wide variety of antigens and must be prepared to detect and discriminate them in order to subsequently drive appropriate responses leading to tolerance or effector responses.
Concerning specifically the presence of DCs in the skin, siting among keratinocytes within the epidermis, dendritic cells named Langerhans cells (LCs) form an extensive network patrolling the whole body surface [32]. These cells do not depend on Flt3L or BATF3 and originally arise first from myeloid precursors derived from the yolk sac during ontogeny, which are later on replaced by monocytes coming from the fetal liver. LCs are described as CD11c+ MHCII+ CD11b+ Langerin+ cells that do not express CD103 or CD8α [32]. In the dermis, two main DC subsets are found: CD11b+ Langerin− dermal DCs and CD103+ CD11b− Langerin+ dermal DCs. Those are equivalent to the conventional or classical DCs described earlier.
LCs have been shown to be necessary and sufficient for the generation of antigen-specific Th17 cells in an experimental model of skin infection with Candida albicans [33]. Moreover, BATF3-dependent CD103+ dermal DCs were found to be major producers of IL-12, being important to drive Th1-mediated responses controlling Leishmania major intradermal infection in mice [34]. In the absence of skin infection or inflammation, migratory LC and dermal CD103+ DCs were shown to efficiently drive the generation of Foxp3+ regulatory T cells when presenting self-antigens to naïve T cells in draining lymph nodes [35]. All these important functions exerted by DCs in the skin could potentially be somehow modulated by the SNS since they can be reached by neurotransmitters released by the fibers innervating the skin.
Similar to what happens to the skin, intestinal mucosa is continuously exposed to diverse antigens, ranging from those related to pathogenic microorganisms to those associated with the diet and commensal microbiota. The ability to differentially respond to these antigens in a tolerogenic or inflammatory way helps to homeostasis maintenance, pathogen elimination, and tissue damage control.
DCs are found throughout the intestine, such as in lamina propria, Peyer’s patches, and intestinal lymphoid follicles, and many DC subtypes are found in different intestinal compartments [36]. Migratory CD103+ CD11b− DCs have been shown to orchestrate tolerogenic responses due to their ability to induce Foxp3+ regulatory T cells [15, 37–39]. IRF8-dependent CD103+ DCs seem also important to support Th1 cell differentiation in mesenteric lymph nodes and to drive Th1-mediated responses upon Trichuris muris infection [40]. Intestinal CD103− CD11b+ DCs were also shown to induce the differentiation of both IFN-γ and IL-17-producing effector T cells [41]. On the opposite, CX3CR1+ DCs are in lamina propria, extend their processes across the epithelial layer into the intestinal lumen and do not migrate to LN after stimulation, being inefficient at priming naïve T cells, and probably have the role of serving as a first line of defense by phagocyting and killing bacteria directly on the epithelial surface [38].
Sympathetic nerve fibers enter the intestine along with arteries and spread throughout myenteric and submucosal plexuses—where cell bodies of neurons from the enteric nervous system are found—and the mucosa. In the gut-associated lymphoid tissue (GALT), it was showed, specifically in Peyer’s patch, that NA fibers enter the serosal surface, cross longitudinally the submucosal border of the muscularis interna, turn radially into an internodular plexus, plunge through the T cell zones, and ramify profusely among lymphocytes, enterochromaffin cells, and plasma cells in the interdomal regions [42]. In the human small intestine, a similar distribution of noradrenergic nerves was found [43].
Although the SNS presence in the intestine is linked to the control of motility, secretion, and also vasoregulation, the presence of NA fibers within the mucosa and the GALT points to an immunomodulatory role [44].
In fact, recently, it has been shown by immunofluorescence and confocal microscopy that a macrophage population in the muscular layer of the gut is in tight contact with sympathetic neurons [45]. Moreover, the authors showed that in the gut, there is heterogeneity in the macrophage population accordingly to the anatomical localization of the cells. Lamina propria macrophages presented a pro-inflammatory profile, while muscularis macrophages presented a more anti-inflammatory profile.
In general, evidence from anatomical and histological studies give support to the notion that in lymphoid organs, skin, and in the gut, dendritic cells and other myeloid cells are likely to have their functions modulated by the sympathetic nervous system.
Norepinephrine production and release
Dopamine, norepinephrine, and epinephrine are catecholamines, a group of chemicals containing a catechol or 3,4-dihydroxyphenyl group and a side chain amine. The synthesis of catecholamines starts with the transformation of the amino acid tyrosine in levodopa through the enzyme TH that is the key rate-limiting enzyme in the biosynthetic pathway of norepinephrine. The levodopa is decarborxylated into dopamine by DOPA decarboxylase. Then, norepinephrine is synthesized from dopamine by dopamine b-hydroxylase and is converted to epinephrine by phenylethanolamine N-methyltransferase. The main source of norepinephrine into lymphoid organs is the sympathetic fibers localized within the spleen and lymph nodes in the T cell zones [42]. Furthermore, increased norepinephrine release in the spleen followed by the activation of antigen-specific T and B cells has been already reported [46], indicating that an adaptive immune response can lead to enhanced sympathetic nervous system activity within lymphoid organs.
Coexistence of mediators in the nervous and the immune systems has long been considered as a universal language of a neuroendocrine-immune modulatory network [47]. In this sense, around the 1990s, the first reports appeared showing that lymphocyte-derived catecholamine could modulate lymphocyte functions in an autocrine and/or paracrine manner [48]. Since then, data have accumulated on the functional relevance of endogenous catecholamine produced by cells of the immune system [49–52]. The presence of catecholamines or TH has been observed in peripheral blood mononuclear cells (PBMCs) [49, 51], bone marrow-derived murine mast cells [53], macrophages [54], and rat lymphocytes [52]. Differently from CD4+ human T cells that are thought to produce significant amounts of catecholamine only after activation [49, 51], CD4+ CD25+ regulatory T cells were found to constitutively express TH and contain substantial amount of dopamine, noradrenaline, and adrenaline [55]. Phagocytes exposed to lipopolysaccharide have been shown to secrete catecholamines, and phagocyte-derived catecholamine was associated with increased inflammatory response in acute lung injury model in vivo [54]. In contrast, an IL-4-stimulated program of alternative activated macrophages was associated with catecholamine secretion required to sustain adaptive thermogenesis [56]. Thus, besides being modulated by the SNS, dendritic cells could also be potentially regulated by catecholamine secreted by other immune cells in a paracrine way.
Adrenergic receptor signaling
In order to respond to the norepinephrine and the epinephrine released either by immune cells or the SNS, dendritic cells and other immune cells must express adrenergic receptors. Adrenergic receptors are seven-transmembrane, G protein-coupled receptors classified according to the type of G protein forming the complex with each receptor and the respective downstream signaling pathway. There are three major types: α1AR, α2AR, and βARs.
So far, three subsets of α1AR have been cloned in humans: α1aAR, α1bAR, and α1dAR, and all of which are coupled to Gαq/11 protein. α1AR stimulation induces phospholipase C activation that mediates hydrolysis of phosphatidylinositol bisphosphate into inositol trisphosphate and diacylglycerol, resulting in increased Ca++ influx [4]. In addition, α1AR stimulation can also activate the mitogen-activated protein kinase (MAPK) family, including the extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs), and p38 kinases.
In turn, α2AR are subdivided in α2aAR, α2bAR, and α2cAR. When stimulated, they all induce activation of the Gαi protein, also known as inhibitory G protein, leading to adenylate cyclase inhibition and, therefore, decreased levels of intracellular cyclic adenosine monophosphate (cAMP). The α2AR plays an important role in the negative feedback loop regulating the release of norepinephrine by sympathetic fibers [57]. Moreover, these receptors are also involved in the release regulation of a number of other neurotransmitters in the central and the peripheral nervous system, such as GABA, dopamine, and serotonin.
Finally, the βAR are divided in β1AR, β2AR, and β3AR. They are all coupled to a stimulatory protein G (Gαs), and their stimulation usually results in an increase of the intracellular cAMP levels via adenylate cyclase activation.
Since cAMP is a second messenger of several different signaling pathways, βAR activation can regulate a diverse array of cellular processes via two major downstream effector systems, which are either dependent on PKA or EPAC.
The canonical βAR signaling pathway involves the cAMP-mediated protein kinase A (PKA) activation, which thereafter phosphorylates specific residues of serine and threonine on target proteins. Thus, PKA regulates a wide variety of cellular processes including gene transcription. Indeed, several transcription factors that are crucial for the immune response can be regulated by PKA-induced phosphorylation, including the cAMP-responsive element binding protein/activating transcription factor (CREB/ATF) family, NFAT, and MAPK. For many years, it was believed that the βAR effects on immune cells were only mediated by PKA activation. Indeed, some studies including data of our own group found that β2AR stimulation controls aspects of CD4+ T cell function in a PKA-dependent manner. On the other hand, in contrast with some studies, we also found that β2AR effects on inhibition of inflammatory cytokine production by dendritic cells occur through PKA-independent signaling pathways.
Alternatively, β2AR stimulation can also activate the guanine exchange proteins directly activated by cAMP (EPAC). EPACs, EPAC-1 and EPAC-2, activate the Ras-like guanine triphosphatase Rap1A [58, 59], which, in turn, stimulates downstream effectors B-Raf, MAP/ERK1/2, and ERK1/2 [60]. EPAC-1 was also shown to regulate gene transcription via the CCAAT enhancer-binding protein (C/EBP) family of transcription factors [61]. Usually, the EPAC-mediated effects on the cells can be distinguished from those mediated by PKA using pharmacologic agonists.
Recently, it has been demonstrated another pathway by which β2AR signaling can regulate the immune response. It involves the family of arrestin proteins and occurs independently of cAMP levels. So far, the arrestin family is divided in β-arrestin-1, β-arrestin-2, arrestin-1, and arrestin-4. For many years, the β-arrestins were considered important only for the internalization and desensitization of ARs (and other GPCRs); however, it is clear now that arrestins are also adaptor molecules, which function as signal transducers to multiple signaling pathways, including MAPKS and nuclear factor-kappaB (NF-κB) [62].
Adrenergic receptors on immune cells
It is well established that innate and adaptive immune cells express both αAR and βAR, being β2AR the most expressed on all immune cells [63, 64]. The AR expression on immune cells varies among the cell types and also according to the degree of cell activation. Several environmental factors present during inflammation can regulate AR expression on immune cells, including TLR ligands, hormones, and cytokines [64–66]. LPS stimulation is related to decreased β2AR expression on macrophages and dendritic cells [64, 66]. In addition, we have recently found that LPS modify the AR expression on dendritic cells, not only decreasing the expression of βAR but also enhancing αAR expression [64]. In T cells, β2AR expression can differ across the subsets of T cells, and this seems to be related, at least in part, to TCR activation and to cytokines present during T cell differentiation. For instance, TCR engagement is associated with a downregulation of β2AR on T cells; thus, naive CD4+ T cells express higher β2AR levels than in vitro activated CD4+ T cells and regulatory FoxP3+ CD4+ T cells [63]. Among the subsets of CD4+ T cells, β2AR expression has been reported on Th1 cell clones [65, 67] and regulatory FoxP3+ T cells [63]; however, it is absent on CD4+ Th2 cells [67] due to IL-4-mediated epigenetic modifications in the b2ar gene [65]. To date, there are no reports describing the expression and function of AR on Th17 cells. Altogether, these data indicate that in general, inflammation changes AR expression on immune cells, inhibiting βAR and enhancing αAR expression, which might be related to the pro-inflammatory process in some immune-mediated diseases. In agreement, an increase of α1AR was found in the inflamed tissue of patients with juvenile idiopathic arthritis [68], while βARs are diminished in rheumatoid arthritis [69].
Modulation of DC function via adrenergic receptor signaling
The SNS through norepinephrine and other neurotransmitters can modulate several aspects of DC function, including migration, antigen uptake, cross-presentation, and mainly cytokine production [64, 70–73]. Regarding dendritic cells in the skin, it was shown that LCs and LC-like cell lines express transcripts for α1AR and β2AR [74]. Treatment of murine epidermal cells with norepinephrine (NE) inhibited antigen presentation in vitro. Accordingly, in vitro pretreatment of epidermal DCs with NE suppressed their ability to present antigen for elicitation of delayed-type hypersensitivity in previously immunized mice. This effect could be blocked using a β2AR antagonist but not a αAR antagonist [74, 75]. The concept that NE can modulate immune responses locally in the skin was further supported by the finding that pharmacological inhibition of NE release by injection of the ganglionic blocker pentolinium led to increased contact hypersensitivity in mice challenged with an antigen in the skin [76]. However, norepinephrine has also been shown to improve the skin DC migration to lymphoid organs [77]. In vivo experiments using the fluorescent molecule FITC as an antigen to induce migration of Langerhans cells to draining lymph nodes as well as skin culture assays showed that NE enhances DC migration via α1AR signaling. Thus, increased DC migration to draining lymph nodes was observed in mice treated systemically with the α2AR antagonist yohimbine, which results in increased NE release due to sympathetic hyperactivity mediated by the blockade of both peripheral and central presynaptic α2ARs. Moreover, local treatment of the skin with the α1AR antagonist prazosin, but not the β2AR antagonist propranolol, inhibited Langerhans cell migration to the draining lymph nodes during sensitization with FITC [77]. However, later on, the same researcher reported that LC migration and the contact hypersensitivity response were increased in mice treated with the β2AR antagonist ICI 118,551 during FITC sensitization [78]. Consistently, they also found that β2AR activation in bone marrow-derived DCs before adoptive transfer could reduce both migration and the contact hypersensitivity response to FITC [78]. The authors suggested that these effects were associated with impaired DC chemotactic response to CCL19 and CCL21 following β2AR activation. Another study fueled the controversy reporting that, due to increased release of NE by the SNS, psychological stress promotes enhanced skin DC migration to lymph nodes, which results in increased antigen-specific CD8+ T cell responses and enhanced skin delayed-type hypersensitivity (DTH) reaction to haptens [79]. It is possible that the SNS and NE differentially influence skin DC migration—and further elicitation of antigen-specific responses—depending on the activation of either α1AR or βAR. In this sense, factors modulating the expression of these receptors in skin DC may be determinant to the final response outcome. In addition, mice injected with intradermal peptidoglycan (PGN) in association with the βAR antagonist propranolol presented increased skin production of pro-inflammatory cytokines such as IL-12 and IL-23 when compared to mice injected with PGN alone. The data indicates a negative modulation of skin inflammation following βAR activation [80]. Furthermore, PGN plus propranolol induced a stronger antigen-specific Th1 recall response and higher DTH response to the same antigen; this βAR-mediated effect seems to be related to a selective increase in the migration of pDCs to the draining lymph nodes [80].
NE has also been shown to play a role in DC endocytosis. Immature and LPS-activated bone marrow-derived dendritic cells (BMDCs) treated with NE display higher capacity to uptake antigens [70]. This NE-mediated effect was shown to rely on α2AR signaling as observed when the BMDCs were pretreated with specific AR antagonists or with a α2AR agonist. Of note, NE markedly activated PI3K and ERK1/2 signaling pathways in DCs; however, NE-induced endocytosis was abrogated when BMDCs were treated with a specific inhibitor of PI3K and LY294002, but not with U0126, a specific inhibitor of MEK1/2, the upstream activator of ERK1/2. Thus, NE enhances antigen uptake by DCs via α2AR-mediated PI3K activation [70].
Following antigen uptake, DCs process and present antigens to T cells. Usually, intracellular antigens are presented by APCs through MHC class I molecules, while extracellular antigens are presented via MHC class II molecules, which then activate CD8+ and CD4 T cells, respectively. Some DCs have the capacity to acquire extracellular antigens, process, and present them via MHC class I for CD8+ T cells, a process called cross-presentation. In vivo, cross-presentation can occur following processing of dead cells, apoptotic bodies, or heat shock protein-associated antigens. Moreover, the cross-presentation pathway is important for tumor-associated antigen presentation or during bacterial or viral infection. It has been shown that β2AR signaling, although did not modify the ability of DC to present exogenous antigens, inhibits in vitro and in vivo cross-presentation by DCs [71]. This effect was shown to be mediated by the inhibitory G protein (Gαi) [71]. Initially, the β2AR was considered to be exclusively associated with Gαs; however, it has become clear that β2AR stimulation may also elicit a different set of signaling pathways, including the activation of MAPK, which is not mediated by Gαs but by the βγ subunits of pertussis toxin-sensitive Gαi [81]. The switching of the coupling of β2AR from Gs to Gi requires PKA-dependent phosphorylation of the receptor [81]. DC exposure to the β2AR agonist salbutamol does not only impair the cross-presentation of exogenous antigens but also affects the endogenous presentation of antigens, as observed when salbutamol-treated DCs infected with a lentivirus expressing the HA antigen displayed lower capability to present the antigen to naive CD8+ T cells as compared with untreated DCs [71]. To better evaluate how β2AR stimulation in DCs could affect antigen cross-presentation, the authors performed in vitro experiments to measure antigen uptake, antigen degradation, and proteasomal activity. They observed that neither antigen uptake, as assessed by flow cytometry using soluble OVA-Alexa 488, or proteasomal activity, addressed using a luminescent assay that measures the chymotrypsin-like, trypsin-like, and caspase-like activities associated with the proteasome complex in cultured cells, were affected following β2AR stimulation in DC. However, using a well-standardized antigen degradation assay that measures OVA degradation at the surface of phagocytosed beads, they observed that phagosomal degradation was partially inhibited after β2AR signaling [71]. In agreement with these data, the ablation of the peripheral SNS in mice using 6-hydroxydopamine (6-OHDA) was shown to enhance the primary CD8+ T cell responses to viral and cellular antigens. Adoptive transfer experiments indicate that enhanced CD8+ T cell responses do not result from permanent alterations in CD8+ T cell function in sympathectomized mice but are rather related to the increased ability of DCs to activate naïve T cells [82]. Thus, β2AR stimulation on DCs impairs antigen cross-presentation by DCs, therefore limiting CD8+ T cell responses. Further studies are necessary to better understand the role of β2AR stimulation in the maintenance of self-tolerance under physiological conditions, controlling T cell activation by DCs as well as its role in providing tumor growth.
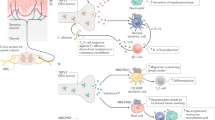
Regarding cytokine secretion, it is well established that NE, via stimulation of β2AR, modulates cytokine production by activated DCs, primarily inhibiting some proinflammatory cytokines, such as TNF-α, IL-12, and IL-6, and increasing the release of IL-10 and IL-33 [64, 71, 72, 83, 84]. β2AR activation was also shown to suppress TLR-9-induced interferon-α secretion by human pDCs. The β2AR-dependent TLR-9 suppression was found to be mediated by bystander cells within peripheral blood mononuclear cells [85]. As expected, β2AR activation in DCs also results in adenylyl cyclase activation, increased cAMP levels, and PKA-dependent CREB phosphorylation [64] (Fig. 1). However, we found that although a PKA inhibitor can totally prevent β2AR-mediated CREB phosphorylation, it was not able to reverse IL-12p70 reduction in LPS-stimulated DC upon β2AR activation [64].

β2 adrenergic receptor (β2AR)-mediated modulation of IL-12/IL-23 production by LPS-stimulated dendritic cells. LPS activation of toll-like receptor 4 (TLR4) promotes IκBα phosphorylation (1). IκBα phosphorylation and further degradation allows p50p65 (NF-κB) translocation to the cell nucleus where it induces IL-12p35 and IL-12p40 transcription. TLR4 signaling also promotes c-JUN phosphorylation, and AP-1 is also thought to favor IL-12p35 and IL-12p40 transcription. Upon β2AR signaling, β-arrestin-1 and β-arrestin-2 (βarr) bind to the IκBα-p50p65 complex and inhibit IκBα phosphorylation, preventing NF-κB translocation to the cell nucleus and thus reducing IL-12p35 and IL-12p40 transcription (2). β2AR activation also blocks LPS-mediated c-JUN phosphorylation, which may contribute to decreased IL-12p35 and IL-12p40 transcription (3). β2AR signaling activates adenylyl cyclase (AC) leading to increased cAMP levels, which, in turn, promote PKA-mediated CREB phosphorylation (4). Alternatively, elevated cAMP levels after β2AR stimulation can also activate EPAC, which induces C/EBP phosphorylation. Phospho-CREB and C/EBP may explain increased IL-23p19 transcription following β2AR activation
A key regulator of the immune response and a target of β2AR signaling is the transcription factor NF-κB. Indeed, NF-κB is a crucial transcription factor for the expression of most of the inflammatory cytokines triggered following TLR stimulation (Fig. 1). For many years, the mechanisms by which β2AR-mediated NF-κB inhibition occurs were unknown. However, recent evidence has arisen demonstrating that β2AR signaling affects important steps of TLR-induced NF-κB activation. That includes downregulation of TLR signaling and inhibition of IkB molecule phosphorylation and degradation in the cytoplasm, leading to an impairment of its function as a transcription factor in the nucleus [64, 86, 87]. Effective TLR4 signaling depends on LPS binding to membrane CD14, that then forms the TLR4/CD14 complex and activates TLR4, which, in turn, activates signal transduction pathways and induces inflammatory gene expression [88] (Fig. 1). Fenoterol, a β2AR agonist, was shown to decrease the membrane-bound TLR4/CD14 complex, due to the redistribution of CD14 on the membrane of the human monocytic cell line THP-1 cells. This effect was dependent on β-arrestin-2, since silencing β-arrestin-2 abolished the redistribution of LPS-induced TLR4/CD14 complex mediated by β2AR [86]. β-arrestin-1 and β-arrestin-2 have long been recognized as important regulators of G protein-coupled receptor signaling, being involved in their internalization and consequent desensitization. More recently, however, it has been reported that β-arrestin-1 and β-arrestin-2 can also directly modulate different intracellular signaling pathways, including NF-κB [62]. Indeed, both β-arrestin-2 and β-arrestin-1 were shown to form a complex with IκBα, which prevents phosphorylation and degradation of IκBα, thus inhibiting NF-κB translocation to the cell nucleus, attenuating the transcription of NF-κB target genes [62, 87, 89] (Fig. 1). Moreover, β2AR stimulation significantly augments the amount of β-arrestin-2 associated with IκBα, which stabilizes IκBα and decreases TNF-α-induced IκBα phosphorylation and degradation, with a consequent reduction in the expression of NF-κB target genes, such as IL-6 and IL-8. Notably, β2AR-mediated enhancement of the β-arrestin-2–IκBα interaction does not seem to be mimicked by forskolin, indicating that this is unlikely to rely on increase in cAMP levels and consequent PKA activation [87]. In agreement with this, we found that β2AR-mediated inhibition of IL-12p70 production by LPS-activated BMDC was associated with decreased IκBα phosphorylation and reduced NF-κBp65 translocation to the nucleus [64] (Fig. 1). Importantly, as mentioned, we demonstrated that impaired IL-12p70 secretion by LPS-stimulated DCs following β2AR activation occurs in a PKA-independent manner. To evaluate whether β-arrestin-2 was involved in the β2AR-mediated downregulation of IL-12 release in LPS-activated DCs, we used BMDC from β-arrestin-2−/− mice. We found that the absence of β-arrestin-2 only partially abrogated the β2AR-mediated IL-12 reduction in LPS-activated DC, indicating that although β2AR stimulation may lead to β-arrestin-2-dependent inhibition of NF-κB activation, additional mechanisms are likely involved [64]. Also, we cannot rule out that possibly, the effect seen on β-arrestin-2−/− DCs was only partial because β-arrestin-1 was also shown to bind to IκBα and inhibit NF-κB activation [89].
Another signaling pathway that we found to be targeted by β2AR stimulation on LPS-stimulated DCs is the JNK pathway [64]. Studies have demonstrated that the activator protein 1 (AP-1) transcription factor has a role in controlling IL-12 production by innate immune cells [90, 91]. In addition, JNK inhibition was shown to decrease IL-12p70 secretion by LPS-stimulated human monocyte-derived DC [92]. Corroborating these findings, we found that JNK inhibition blocked LPS-induced c-Jun phosphorylation and reduced IL-12p70 secretion by DC, supporting the idea that AP-1 is an important transcription factor promoting IL-12p70 expression after LPS stimulation in DC [64] (Fig. 1). Moreover, β2AR signaling inhibited AP-1 transcription factor activation, blocking c-Jun phosphorylation in LPS-stimulated DC. Interestingly, β2AR-mediated blockade of c-Jun phosphorylation was found to be PKA independent, similar to what was verified for β2AR-mediated decrease in IL-12 secretion upon LPS challenge [64] (Fig. 1). The mechanisms by which β2AR activation blocks c-Jun phosphorylation are still to be determined. Altogether, gathered data indicate that β2AR activation suppresses NF-κB and JNK signaling pathways, which are important to elicit IL-12 secretion by LPS-stimulated DC.
Despite significantly reducing the amount of IL-12p70 secreted by LPS-stimulated DCs, β2AR activation did not alter that much the production of LPS-induced IL-23, another cytokine member of the IL-12 family [64]. This may be considered an intriguing finding since TLR-induced IL-23p19 gene expression was also found to be dependent on NF-κB and AP-1 transcription factors [93, 94]. Actually, we found that, in contrast to decreased levels of IL-12p35 transcripts, β2AR activation induced an increase in LPS-induced IL-23p19 transcript levels in DCs [64]. One possible explanation for this paradox could be that, even though compromising NF-κB and AP-1, β2AR signaling on LPS-stimulated DC is perhaps activating other transcription factors that are positive regulators of IL-23 expression. In this sense, it was shown before that the IL-23p19 promoter region possesses binding sites for CREB and C/AATT enhancer-binding protein β (C/EBP). C/EBP can be phosphorylated via EPAC, which, in turn, is activated by cAMP (Fig. 1). Indeed, EPAC (via C/EBP) and phospho-CREB were shown to induce p19 transcriptional activity [95]. Accordingly, we found that a specific EPAC activator (8-CPT-2Me-cAMP) led to increased IL-23 secretion by LPS-stimulated DC [64]. Therefore, phospho-CREB and/or EPAC activation by increased cAMP levels following β2AR stimulation can possibly explain the augmented LPS-induced p19 transcription in DC.
Since it diminishes LPS-induced IL-12p70 while did not modify IL-23 secretion by DCs, β2AR activation promotes a shift in IL-12p70/IL-23 ratio. Hence, LPS-stimulated DCs usually secrete twice as much IL-12p70 as IL-23. Following β2AR signaling, DCs produce twice as much IL-23 as IL-12p70 [64]. Matching the profile of cytokines produced by DCs upon recognition of pathogenic agents with the generation of the appropriate CD4 T cell response is crucial to efficiently control each type of infection faced by the host. Accordingly, we found that the β2AR-mediated change in the profile of cytokines produced by LPS-stimulated DCs impacts the generation of adaptive immune responses. Indeed, β2AR stimulation on DCs later on stimulated with LPS led to CD4+ T cells that upon TCR engagement produced less IFN-γ and more IL-17 [64]. Thus, β2AR signaling in DCs changes the way these cells sense LPS, modifying the profile of cytokines being produced. This impacts adaptive immune responses favoring the development of Th17 cells and impairing the generation of Th1 cells. Of note, the β2AR-mediated changes in the profile of cytokines produced by LPS-stimulated DCs also include increased IL-10 production, an important anti-inflammatory cytokine [64]. How this particular effect impacts the way DCs drive adaptive immune responses is still to be unravel.
Concluding remarks
Signaling via adrenergic receptors in dendritic cells can modulate many of their functions, including migration, antigen uptake, antigen presentation, and cytokine production. This modulation may differentially impact immune-mediated diseases depending on the dendritic cell subset being targeted and also on the context of the regulation exerted by neurotransmitters released by the SNS. For instance, pDCs in the skin, through interferon-α production, have been implicated in the pathophysiology of inflammatory diseases such as psoriasis [96]. Downregulation of βAR expression/function has been reported in the skin lesions from patients with psoriasis [97]. The lack of regulation by the SNS of immune cells within the skin could be one of the mechanisms favoring the disease [98]. On the other hand, since type I interferons play an important role in anti-viral immunity, the PKA-dependent adrenergic-mediated inhibition of interferon-α production by pDCs was shown to favor HIV-1 replication [99]. In addition, house dust mite-induced allergic airway inflammation was found to be attenuated by a β2AR agonist through modulation of DC function [100]. Treatment with salbutamol, a β2AR agonist, ameliorated the severity of adjuvant-induced arthritis in mice and rats [101, 102], and β2AR signaling reduction in DCs was shown to be involved in the inflammatory response of arthritic rats [102]. Besides the reduced expression of β2AR in DCs, the development of arthritis was also associated with a progressive loss of the sympathetic innervation within the synovial tissue [103, 104]. A similar loss in the density of sympathetic innervation was found to occur in intestinal inflammatory disorders as well [44]. Recently, it was found that NE released by sympathetic fibers upon intestinal infection drives a tissue-protective transcriptional program in myeloid cells via β2AR signaling [45]. β2AR activation in myeloid cells present in the muscularis, which are in very close contact with neuronal cells within the myenteric plexus, induces the expression of a gene profile resembling that present in alternatively activated macrophages [45]. It was suggested that this tissue-specific regulation of myeloid cells by the SNS would be of particular importance to avoid neuronal damage in the myenteric plexus upon infection. Indeed, previous findings indicate that inflammation and neuronal degeneration in the myenteric plexus are involved in the pathogenesis of intestinal inflammatory diseases such as the irritable bowel syndrome [105].
We are still learning about the modulation of DC functions by the SNS, and further studies are necessary to understand how this particular neuroimmune interaction can affect distinct immune-mediated diseases or help in homeostasis maintenance.
References
Veiga-Fernandes H, Mucida D (2016) Neuro-immune interactions at barrier surfaces. Cell 165(4):801–811. doi:10.1016/j.cell.2016.04.041
Costa-Pinto FA, Basso AS, De Sa-Rocha LC, Britto LR, Russo M, Palermo-Neto J (2006) Neural correlates of IgE-mediated allergy. Ann N Y Acad Sci 1088:116–131. doi:10.1196/annals.1366.028
Sternberg EM (2006) Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol 6(4):318–328. doi:10.1038/nri1810
Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES (2000) The sympathetic nerve—an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev 52(4):595–638
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421(6921):384–388. doi:10.1038/nature01339
Rosas-Ballina M, Olofsson PS, Ochani M, Valdés-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, Mak TW, Tracey KJ (2011) Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334(6052):98–101. doi:10.1126/science.1209985
Mildner A, Jung S (2014) Development and function of dendritic cell subsets. Immunity 40(5):642–656. doi:10.1016/j.immuni.2014.04.016
Satpathy AT, Wu X, Albring JC, Murphy KM (2012) Re(de)fining the dendritic cell lineage. Nat Immunol 13(12):1145–1154. doi:10.1038/ni.2467
Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG (2007) Identification of clonogenic common Flt3 + M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol 8(11):1207–1216. doi:10.1038/ni1518
Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, Carotta S, O'Keeffe M, Bahlo M, Papenfuss A, Kwak JY, Wu L, Shortman K (2007) Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol 8(11):1217–1226. doi:10.1038/ni1522
Liu K, Waskow C, Liu X, Yao K, Hoh J, Nussenzweig M (2007) Origin of dendritic cells in peripheral lymphoid organs of mice. Nat Immunol 8(6):578–583. doi:10.1038/ni1462
Roses RE, Xu S, Xu M, Koldovsky U, Koski G, Czerniecki BJ (2008) Differential production of IL-23 and IL-12 by myeloid-derived dendritic cells in response to TLR agonists. J Immunol 181(7):5120–5127
Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A (2005) Selected toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol 6(8):769–776. doi:10.1038/ni1223
Osorio F, LeibundGut-Landmann S, Lochner M, Lahl K, Sparwasser T, Eberl G, Reis e Sousa C (2008) DC activated via dectin-1 convert Treg into IL-17 producers. Eur J Immunol 38(12):3274–3281. doi:10.1002/eji.200838950
Esterházy D, Loschko J, London M, Jove V, Oliveira TY, Mucida D (2016) Classical dendritic cells are required for dietary antigen-mediated induction of peripheral Treg cells and tolerance. Nat Immunol 17(5):545–555. doi:10.1038/ni.3408
Nance DM, Hopkins DA, Bieger D (1987) Re-investigation of the innervation of the thymus gland in mice and rats. Brain Behav Immun 1(2):134–147
Trotter RN, Stornetta RL, Guyenet PG, Roberts MR (2007) Transneuronal mapping of the CNS network controlling sympathetic outflow to the rat thymus. Auton Neurosci 131(1–2):9–20. doi:10.1016/j.autneu.2006.06.001
Nance DM, Burns J (1989) Innervation of the spleen in the rat: evidence for absence of afferent innervation. Brain Behav Immun 3(4):281–290
Romeo HE, Fink T, Yanaihara N, Weihe E (1994) Distribution and relative proportions of neuropeptide Y- and proenkephalin-containing noradrenergic neurones in rat superior cervical ganglion: separate projections to submaxillary lymph nodes. Peptides 15(8):1479–1487
Nance DM, Sanders VM (2007) Autonomic innervation and regulation of the immune system (1987-2007. Brain Behav Immun 21(6):736–745
Kurkowski R, Kummer W, Heym C (1990) Substance P-immunoreactive nerve fibers in tracheobronchial lymph nodes of the Guinea pig: origin, ultrastructure and coexistence with other peptides. Peptides 11(1):13–20
Madden KS, Sanders VM, Felten DL (1995) Catecholamine influences and sympathetic neural modulation of immune responsiveness. Annu Rev Pharmacol Toxicol 35:417–448
Schafer MK, Eiden LE, Weihe E (1998) Cholinergic neurons and terminal fields revealed by immunohistochemistry for the vesicular acetylcholine transporter. II. The peripheral nervous system. Neuroscience 84(2):361–376
Bellinger DL, Lorton D, Hamill RW, Felten SY, Felten DL (1993) Acetylcholinesterase staining and choline acetyltransferase activity in the young adult rat spleen: lack of evidence for cholinergic innervation. Brain Behav Immun 7(3):191–204
Bellinger DL, Felten SY, Lorton D, Felten DL (1989) Origin of noradrenergic innervation of the spleen in rats. Brain Behav Immun 3(4):291–311
Meltzer JC, Grimm PC, Greenberg AH, Nance DM (1997) Enhanced immunohistochemical detection of autonomic nerve fibers, cytokines and inducible nitric oxide synthase by light and fluorescent microscopy in rat spleen. J Histochem Cytochem 45(4):599–610
Cesta MF (2006) Normal structure, function, and histology of the spleen. Toxicol Pathol 34(5):455–465. doi:10.1080/01926230600867743
Demonceau C, Marshall AS, Sales J, Heinen E (2008) Investigation of close interactions between sympathetic neural fibres and the follicular dendritic cells network in the mouse spleen. Eur J Histochem 52(2):85–92
Rice FL, Albrecht PJ, Wymer JP, Black JA, Merkies IS, Faber CG, Waxman SG (2015) Sodium channel Nav1.7 in vascular myocytes, endothelium, and innervating axons in human skin. Mol Pain 11:26. doi:10.1186/s12990-015-0024-3
Botchkarev VA, Peters EM, Botchkareva NV, Maurer M, Paus R (1999) Hair cycle-dependent changes in adrenergic skin innervation, and hair growth modulation by adrenergic drugs. J Invest Dermatol 113(6):878–887. doi:10.1046/j.1523-1747.1999.00791.x
Falck B, Rorsman H (1963) Observation on the adrenergic innervation of the skin. Experientia 19:2
Clausen BE, Stoitzner P (2015) Functional specialization of skin dendritic cell subsets in regulating T cell responses. Front Immunol 6:534. doi:10.3389/fimmu.2015.00534
Igyártó BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, Edelson BT, Zurawski SM, Malissen B, Zurawski G, Berman J, Kaplan DH (2011) Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity 35(2):260–272. doi:10.1016/j.immuni.2011.06.005
Martínez-López M, Iborra S, Conde-Garrosa R, Sancho D (2015) Batf3-dependent CD103+ dendritic cells are major producers of IL-12 that drive local Th1 immunity against Leishmania major infection in mice. Eur J Immunol 45(1):119–129. doi:10.1002/eji.201444651
Idoyaga J, Fiorese C, Zbytnuik L, Lubkin A, Miller J, Malissen B, Mucida D, Merad M, Steinman RM (2013) Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest 123(2):844–854. doi:10.1172/jci65260
Gross M, Salame TM, Jung S (2015) Guardians of the gut—murine intestinal macrophages and dendritic cells. Front Immunol 6:254. doi:10.3389/fimmu.2015.00254
Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F (2007) A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 204(8):1757–1764. doi:10.1084/jem.20070590
Schulz O, Jaensson E, Persson EK, Liu X, Worbs T, Agace WW, Pabst O (2009) Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J Exp Med 206(13):3101–3114. doi:10.1084/jem.20091925
Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y (2007) Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 204(8):1775–1785. doi:10.1084/jem.20070602
Luda KM, Joeris T, Persson EK, Rivollier A, Demiri M, Sitnik KM, Pool L, Holm JB, Melo-Gonzalez F, Richter L, Lambrecht BN, Kristiansen K, Travis MA, Svensson-Frej M, Kotarsky K, Agace WW (2016) IRF8 transcription-factor-dependent classical dendritic cells are essential for intestinal T cell homeostasis. Immunity 44(4):860–874. doi:10.1016/j.immuni.2016.02.008
Cerovic V, Houston SA, Scott CL, Aumeunier A, Yrlid U, Mowat AM, Milling SW (2013) Intestinal CD103(−) dendritic cells migrate in lymph and prime effector T cells. Mucosal Immunol 6(1):104–113. doi:10.1038/mi.2012.53
Felten DL, Felten SY, Carlson SL, Olschowka JA, Livnat S (1985) Noradrenergic and peptidergic innervation of lymphoid tissue. J Immunol 135(2 Suppl):755s–765s
Llewellyn-Smith IJ, Furness JB, O'Brien PE, Costa M (1984) Noradrenergic nerves in human small intestine. Distribution and ultrastructure. Gastroenterology 87(3):513–529
Straub RH, Wiest R, Strauch UG, Härle P, Schölmerich J (2006) The role of the sympathetic nervous system in intestinal inflammation. Gut 55(11):1640–1649. doi:10.1136/gut.2006.091322
Gabanyi I, Muller PA, Feighery L, Oliveira TY, Costa-Pinto FA, Mucida D (2016) Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell 164(3):378–391. doi:10.1016/j.cell.2015.12.023
Leonard JP, MacKenzie FJ, Patel HA, Cuzner ML (1991) Hypothalamic noradrenergic pathways exert an influence on neuroendocrine and clinical status in experimental autoimmune encephalomyelitis. Brain Behav Immun 5(4):328–338
Blalock JE (2005) The immune system as the sixth sense. J Intern Med 257(2):126–138. doi:10.1111/j.1365-2796.2004.01441.x
Bergquist J, Tarkowski A, Ekman R, Ewing A (1994) Discovery of endogenous catecholamines in lymphocytes and evidence for catecholamine regulation of lymphocyte function via an autocrine loop. Proc Natl Acad Sci U S A 91(26):12912–12916
Cosentino M, Marino F, Bombelli R, Ferrari M, Rasini E, Lecchini S, Frigo G (2002) Stimulation with phytohaemagglutinin induces the synthesis of catecholamines in human peripheral blood mononuclear cells: role of protein kinase C and contribution of intracellular calcium. J Neuroimmunol 125(1–2):125–133
Cosentino M, Marino F, Bombelli R, Ferrari M, Lecchini S, Frigo G (2003) Unravelling dopamine (and catecholamine) physiopharmacology in lymphocytes: open questions. Trends Immunol 24(11):581–582 author reply 582-583
Cosentino M, Zaffaroni M, Ferrari M, Marino F, Bombelli R, Rasini E, Frigo G, Ghezzi A, Comi G, Lecchini S (2005) Interferon-gamma and interferon-beta affect endogenous catecholamines in human peripheral blood mononuclear cells: implications for multiple sclerosis. J Neuroimmunol 162(1–2):112–121. doi:10.1016/j.jneuroim.2005.01.019
Qiu YH, Cheng C, Dai L, Peng YP (2005) Effect of endogenous catecholamines in lymphocytes on lymphocyte function. J Neuroimmunol 167(1–2):45–52. doi:10.1016/j.jneuroim.2005.06.007
Freeman JG, Ryan JJ, Shelburne CP, Bailey DP, Bouton LA, Narasimhachari N, Domen J, Siméon N, Couderc F, Stewart JK (2001) Catecholamines in murine bone marrow derived mast cells. J Neuroimmunol 119(2):231–238
Flierl MA, Rittirsch D, Nadeau BA, Chen AJ, Sarma JV, Zetoune FS, McGuire SR, List RP, Day DE, Hoesel LM, Gao H, Van Rooijen N, Huber-Lang MS, Neubig RR, Ward PA (2007) Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature 449(7163):721–725. doi:10.1038/nature06185
Cosentino M, Fietta AM, Ferrari M, Rasini E, Bombelli R, Carcano E, Saporiti F, Meloni F, Marino F, Lecchini S (2007) Human CD4 + CD25+ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine/paracrine inhibitory functional loop. Blood 109(2):632–642. doi:10.1182/blood-2006-01-028423
Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A (2011) Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480(7375):104–108. doi:10.1038/nature10653
Hein L, Altman JD, Kobilka BK (1999) Two functionally distinct alpha2-adrenergic receptors regulate sympathetic neurotransmission. Nature 402(6758):181–184. doi:10.1038/46040
de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396(6710):474–477. doi:10.1038/24884
Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM (1998) A family of cAMP-binding proteins that directly activate Rap1. Science 282(5397):2275–2279
Borland G, Smith BO, Yarwood SJ (2009) EPAC proteins transduce diverse cellular actions of cAMP. Br J Pharmacol 158(1):70–86. doi:10.1111/j.1476-5381.2008.00087.x
Yarwood SJ, Borland G, Sands WA, Palmer TM (2008) Identification of CCAAT/enhancer-binding proteins as exchange protein activated by cAMP-activated transcription factors that mediate the induction of the SOCS-3 gene. J Biol Chem 283(11):6843–6853. doi:10.1074/jbc.M710342200
Ma L, Pei G (2007) Beta-arrestin signaling and regulation of transcription. J Cell Sci 120(Pt 2):213–218. doi:10.1242/jcs.03338
Guereschi MG, Araujo LP, Maricato JT, Takenaka MC, Nascimento VM, Vivanco BC, Reis VO, Keller AC, Brum PC, Basso AS (2013) Beta2-adrenergic receptor signaling in CD4(+) Foxp3(+) regulatory T cells enhances their suppressive function in a PKA-dependent manner. Eur J Immunol 43(4):1001–1012. doi:10.1002/eji.201243005
Takenaka MC, Araujo LP, Maricato JT, Nascimento VM, Guereschi MG, Rezende RM, Quintana FJ, Basso AS (2016) Norepinephrine controls effector T cell differentiation through β2-adrenergic receptor-mediated inhibition of NF-κB and AP-1 in dendritic cells. J Immunol 196(2):637–644. doi:10.4049/jimmunol.1501206
McAlees JW, Smith LT, Erbe RS, Jarjoura D, Ponzio NM, Sanders VM (2011) Epigenetic regulation of beta2-adrenergic receptor expression in T(H)1 and T(H)2 cells. Brain Behav Immun 25(3):408–415. doi:10.1016/j.bbi.2010.10.019
Kizaki T, Izawa T, Sakurai T, Haga S, Taniguchi N, Tajiri H, Watanabe K, Day NK, Toba K, Ohno H (2008) Beta2-adrenergic receptor regulates toll-like receptor-4-induced nuclear factor-kappaB activation through beta-arrestin 2. Immunology 124(3):348–356. doi:10.1111/j.1365-2567.2007.02781.x
Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE (1997) Differential expression of the beta2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help. J Immunol 158(9):4200–4210
Heijnen CJ, Rouppe van der Voort C, Wulffraat N, van der Net J, Kuis W, Kavelaars A (1996) Functional alpha 1-adrenergic receptors on leukocytes of patients with polyarticular juvenile rheumatoid arthritis. J Neuroimmunol 71(1–2):223–226
Baerwald C, Graefe C, von Wichert P, Krause A (1992) Decreased density of beta-adrenergic receptors on peripheral blood mononuclear cells in patients with rheumatoid arthritis. J Rheumatol 19(2):204–210
Yanagawa Y, Matsumoto M, Togashi H (2010) Enhanced dendritic cell antigen uptake via alpha2 adrenoceptor-mediated PI3K activation following brief exposure to noradrenaline. J Immunol 185(10):5762–5768. doi:10.4049/jimmunol.1001899
Hervé J, Dubreil L, Tardif V, Terme M, Pogu S, Anegon I, Rozec B, Gauthier C, Bach JM, Blancou P (2013) β2-Adrenoreceptor agonist inhibits antigen cross-presentation by dendritic cells. J Immunol 190(7):3163–3171. doi:10.4049/jimmunol.1201391
Manni M, Granstein RD, Maestroni G (2011) β2-adrenergic agonists bias TLR-2 and NOD2 activated dendritic cells towards inducing an IL-17 immune response. Cytokine 55(3):380–386. doi:10.1016/j.cyto.2011.05.013
Nijhuis LE, Olivier BJ, Dhawan S, Hilbers FW, Boon L, Wolkers MC, Samsom JN, de Jonge WJ (2014) Adrenergic beta2 receptor activation stimulates anti-inflammatory properties of dendritic cells in vitro. PLoS One 9(1):e85086. doi:10.1371/journal.pone.0085086
Seiffert K, Hosoi J, Torii H, Ozawa H, Ding W, Campton K, Wagner JA, Granstein RD (2002) Catecholamines inhibit the antigen-presenting capability of epidermal Langerhans cells. J Immunol 168(12):6128–6135
Seiffert K, Granstein RD (2006) Neuroendocrine regulation of skin dendritic cells. Ann N Y Acad Sci 1088:195–206. doi:10.1196/annals.1366.011
Maestroni GJ (2002) Short exposure of maturing, bone marrow-derived dendritic cells to norepinephrine: impact on kinetics of cytokine production and Th development. J Neuroimmunol 129(1–2):106–114
Maestroni GJ (2000) Dendritic cell migration controlled by alpha 1b-adrenergic receptors. J Immunol 165(12):6743–6747
Maestroni GJ, Mazzola P (2003) Langerhans cells beta 2-adrenoceptors: role in migration, cytokine production, Th priming and contact hypersensitivity. J Neuroimmunol 144(1–2):91–99
Saint-Mezard P, Chavagnac C, Bosset S, Ionescu M, Peyron E, Kaiserlian D, Nicolas JF, Bérard F (2003) Psychological stress exerts an adjuvant effect on skin dendritic cell functions in vivo. J Immunol 171(8):4073–4080
Manni M, Maestroni GJ (2008) Sympathetic nervous modulation of the skin innate and adaptive immune response to peptidoglycan but not lipopolysaccharide: involvement of beta-adrenoceptors and relevance in inflammatory diseases. Brain Behav Immun 22(1):80–88. doi:10.1016/j.bbi.2007.06.016
Daaka Y, Luttrell LM, Lefkowitz RJ (1997) Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase a. Nature 390(6655):88–91. doi:10.1038/36362
Grebe KM, Hickman HD, Irvine KR, Takeda K, Bennink JR, Yewdell JW (2009) Sympathetic nervous system control of anti-influenza CD8+ T cell responses. Proc Natl Acad Sci U S A 106(13):5300–5305. doi:10.1073/pnas.0808851106
Yanagawa Y, Matsumoto M, Togashi H (2011) Adrenoceptor-mediated enhancement of interleukin-33 production by dendritic cells. Brain Behav Immun 25(7):1427–1433. doi:10.1016/j.bbi.2011.04.012
Haskó G, Szabó C, Németh ZH, Salzman AL, Vizi ES (1998) Stimulation of beta-adrenoceptors inhibits endotoxin-induced IL-12 production in normal and IL-10 deficient mice. J Neuroimmunol 88(1–2):57–61
Hilbert T, Bongartz J, Weisheit C, Knüfermann P, Baumgarten G, Hoeft A, Poth JM (2013) Beta2-adrenoceptor stimulation suppresses TLR9-dependent IFNA1 secretion in human peripheral blood mononuclear cells. PLoS One 8(5):e65024. doi:10.1371/journal.pone.0065024
Wang W, Xu M, Zhang YY, He B (2009) Fenoterol, a beta(2)-adrenoceptor agonist, inhibits LPS-induced membrane-bound CD14, TLR4/CD14 complex, and inflammatory cytokines production through beta-arrestin-2 in THP-1 cell line. Acta Pharmacol Sin 30(11):1522–1528. doi:10.1038/aps.2009.153
Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, Pei G (2004) Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol Cell 14(3):303–317
Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2(8):675–680. doi:10.1038/90609
Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ (2004) Beta-arrestin inhibits NF-kappaB activity by means of its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc Natl Acad Sci U S A 101(23):8603–8607. doi:10.1073/pnas.0402851101
Zhu C, Gagnidze K, Gemberling JH, Plevy SE (2001) Characterization of an activation protein-1-binding site in the murine interleukin-12 p40 promoter. Demonstration of novel functional elements by a reductionist approach. J Biol Chem 276(21):18519–18528. doi:10.1074/jbc.M100440200
Agrawal S, Agrawal A, Doughty B, Gerwitz A, Blenis J, Van Dyke T, Pulendran B (2003) Cutting edge: different toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J Immunol 171(10):4984–4989
Nakahara T, Uchi H, Urabe K, Chen Q, Furue M, Moroi Y (2004) Role of c-Jun N-terminal kinase on lipopolysaccharide induced maturation of human monocyte-derived dendritic cells. Int Immunol 16(12):1701–1709. doi:10.1093/intimm/dxh171
Carmody RJ, Ruan Q, Liou HC, Chen YH (2007) Essential roles of c-Rel in TLR-induced IL-23 p19 gene expression in dendritic cells. J Immunol 178(1):186–191
Liu W, Ouyang X, Yang J, Liu J, Li Q, Gu Y, Fukata M, Lin T, He JC, Abreu M, Unkeless JC, Mayer L, Xiong H (2009) AP-1 activated by toll-like receptors regulates expression of IL-23 p19. J Biol Chem 284(36):24006–24016. doi:10.1074/jbc.M109.025528
Kocieda VP, Adhikary S, Emig F, Yen JH, Toscano MG, Ganea D (2012) Prostaglandin E2-induced IL-23p19 subunit is regulated by cAMP-responsive element-binding protein and C/AATT enhancer-binding protein β in bone marrow-derived dendritic cells. J Biol Chem 287(44):36922–36935. doi:10.1074/jbc.M112.402958
Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, Burg G, Liu YJ, Gilliet M (2005) Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med 202(1):135–143. doi:10.1084/jem.20050500
Steinkraus V, Steinfath M, Stöve L, Körner C, Abeck D, Mensing H (1993) Beta-adrenergic receptors in psoriasis: evidence for down-regulation in lesional skin. Arch Dermatol Res 285(5):300–304
Halevy S, Livni E (1993) Beta-adrenergic blocking drugs and psoriasis: the role of an immunologic mechanism. J Am Acad Dermatol 29(3):504–505
Collado-Hidalgo A, Sung C, Cole S (2006) Adrenergic inhibition of innate anti-viral response: PKA blockade of type I interferon gene transcription mediates catecholamine support for HIV-1 replication. Brain Behav Immun 20(6):552–563. doi:10.1016/j.bbi.2006.01.005
Kato G, Takahashi K, Tashiro H, Kurata K, Shirai H, Kimura S, Hayashi S (2014) β2 adrenergic agonist attenuates house dust mite-induced allergic airway inflammation through dendritic cells. BMC Immunol 15:39. doi:10.1186/s12865-014-0039-y
Malfait AM, Malik AS, Marinova-Mutafchieva L, Butler DM, Maini RN, Feldmann M (1999) The beta2-adrenergic agonist salbutamol is a potent suppressor of established collagen-induced arthritis: mechanisms of action. J Immunol 162(10):6278–6283
Wu H, Chen J, Song S, Yuan P, Liu L, Zhang Y, Zhou A, Chang Y, Zhang L, Wei W (2016) Beta2-adrenoceptor signaling reduction in dendritic cells is involved in the inflammatory response in adjuvant-induced arthritic rats. Sci Rep 6:24548. doi:10.1038/srep24548
Miller LE, Jüsten HP, Schölmerich J, Straub RH (2000) The loss of sympathetic nerve fibers in the synovial tissue of patients with rheumatoid arthritis is accompanied by increased norepinephrine release from synovial macrophages. FASEB J 14(13):2097–2107. doi:10.1096/fj.99-1082com
Weidler C, Holzer C, Harbuz M, Hofbauer R, Angele P, Schölmerich J, Straub RH (2005) Low density of sympathetic nerve fibres and increased density of brain derived neurotrophic factor positive cells in RA synovium. Ann Rheum Dis 64(1):13–20. doi:10.1136/ard.2003.016154
Törnblom H, Lindberg G, Nyberg B, Veress B (2002) Full-thickness biopsy of the jejunum reveals inflammation and enteric neuropathy in irritable bowel syndrome. Gastroenterology 123(6):1972–1979. doi:10.1053/gast.2002.37059
Acknowledgments
Research in the Basso Laboratory is supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (grants 08/58564-9 and 14/24156-2) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (grants 475947/2013-4 and 400450/2014-3). M.G.G. is supported by FAPESP (grant 2013/13110-9).
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Dendritic Cell Subsets and Immune-mediated Diseases - Guest Editor: Francisco Quintana
Rights and permissions
About this article

Cite this article
Takenaka, M.C., Guereschi, M.G. & Basso, A.S. Neuroimmune interactions: dendritic cell modulation by the sympathetic nervous system. Semin Immunopathol 39, 165–176 (2017). https://doi.org/10.1007/s00281-016-0590-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-016-0590-0