
Abstract
Dendritic cells (DC) are unique hematopoietic cells, linking innate and adaptive immune responses. In particular, they are considered as the most potent antigen presenting cells, governing both T cell immunity and tolerance. In view of their exceptional ability to present antigen and to interact with T cells, DC play distinct roles in shaping T cell development, differentiation and function. The outcome of the DC-T cell interaction is determined by the state of DC maturation, the type of DC subset, the cytokine microenvironment and the tissue location. Both regulatory T cells (Tregs) and DC are indispensable for maintaining central and peripheral tolerance. Over the past decade, accumulating data indicate that DC critically contribute to Treg differentiation and homeostasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Dendritic cells (DC) serve as unique sentinels of the immune system, continuously sampling their environment and exerting different properties that in turn determine immunological outcomes. Although DC do not serve as effector cells that fight against pathogens, they control adaptive immunity by providing essential signals that are mandatory for directing the desired immune response. Apart from antigen presentation, DC deliver co-stimulatory signals and produce cytokines, which are necessary for instructing appropriate effector or regulatory T cell responses. In this review, we will examine different aspects of DC-derived tolerance.
DC lineage and subsets
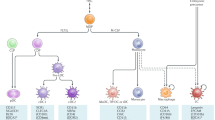
All DC develop from a common macrophage/dendritic cell progenitor (MDP) found in the bone marrow (BM) [1], which further proceeds to differentiate to the monocyte/macrophage lineage or to the common dendritic cell progenitor (CDP) [2, 3]. CDP in the BM give rise to both plasmacytoid DC (pDC) and pre-DC progenitors [4–6]. pDC complete their last step of maturation in the BM, before they egress into the blood stream as mature functional cells. Pre-DC, on the other hand, migrate through the vascular system to their final residence in the tissues or lymphoid organs, where they finish their differentiation into distinct conventional DC subsets, namely, CD8α+/CD103+ DC or CD11b+ DC [7]. DC differentiation, lineage and subsets are summarized in Fig. 1.

Dendritic cell development. Macrophage dendritic cell progenitors (MDP) give rise to either monocytes or common dendritic cell progenitors (CDP). Dependent on macrophage colony stimulating factor (M-CSF) or granulocyte macrophage colony stimulating factor (GM-CSF), monocytes develop into macrophages or monocyte-derived DC, respectively. Monocyte-derived DC give rise to CD11b+ ESAMlow cells. CDP are developing into pDC in a manner dependent on Flt3L, PU.1, E2–2, Ikaros and IRF8. CDP also give rise to pre-DC, which are the progenitors of the conventional CD11b+ ESAMhigh, CD8α+ and CD103+ dendritic cells. The development of CD11b+ ESAMhigh DC is dependent on IRF2, IRF4, Notch2, RelB and LTβ, while the development of both CD8α+ and CD103+ DC is dependent on Flt3L, ID2, IRF8, Batf3 and NFIL3
DC represent a heterogeneous family of myeloid antigen presenting cells (APCs) composed of several different subsets. They are found throughout the entire body, and although 2–4 % of all leukocytes are DC in any given tissue, the composition of the different subsets varies between organs. These subsets are defined by their localization, expression of surface proteins and functionality. Typically, DC are categorized into two distinct classes: pDC, which produce large quantities of type I interferon in response to viral infection and conventional DC, which are highly potent APCs, in particular, for activating naïve T cells. Among the conventional DC, CD8α+ DC are important for orchestrating immune responses against intracellular pathogens, whereas CD11b+ DC are thought to be more relevant for fighting extracellular pathogens.
pDC
pDC are defined as CD11clow MHC-IIlow B220+ PDCA-1+ Siglec-H+ cells. Their development is dependent on E2-2 and IRF8 [8, 9]. pDC are specialized in high type I interferon secretion [10–12]. Steady state pDC are poor APCs, although following their activation by pathogens they are fully capable of priming naïve T cells [9]. Freshly isolated human pDC induce anergy of human CD4+ T cell clones [13, 14]. Similarly, freshly isolated mouse antigen-pulsed splenic pDC induce antigen-specific T cell anergy [15, 16]. Naive T cell stimulation using CpG oligonucleotide-stimulated mesenteric LN-derived pDC gave rise to Tregs with suppressive activity [17]. In vitro human pDC induce T cell differentiation into IL-10-producing Tregs [14, 18]. Taken together, these data indicate that both mouse and human pDC induce CD4+CD25+ FOXP3+ regulatory T cells (Tregs).
CD8α+ and CD103+ DC
Development of both lymphoid organ-resident CD8α+ DC and their tissue-resident CD103+ equivalents was shown to be dependent on tyrosine kinase receptor fms-like tyrosine kinase 3 ligand (Flt3L) [19, 20], inhibitor of DNA binding protein 2 (ID2) [5, 21, 22], the transcription factor interferon regulatory factor 8 (IRF8) [23, 24] and the basic leucine zipper transcription factor ATF-like3 (Batf3) [25, 26]. The CD8α+ DC participate in central as well as peripheral tolerance induction due to their ability to produce high amounts of TGFβ in the steady state [27]. Furthermore, they can induce peripheral tolerance towards tissue-associated self-antigens also via direct contact with self-reactive T cells, which lead to the death of the latter [28, 29]. Their tolerogenic potential also becomes apparent when targeting antigen to CD205 (DEC205), which induces clonal deletion [30, 31] and Treg differentiation [32]. Moreover, a recent publication using conditional ablation of DEC205+ DC established the importance of these cells in the generation of thymic Tregs as well as the maintenance of Tregs in the periphery, i.e. mucosal tissues [33].
CD11b+ DC
CD11b+ DC development is dependent on Flt3L, Notch2, IRF2, IRF4, RelB, LTβ and GM-CSF [34, 35]. These cells are found in both lymphoid organs and peripheral tissues. They are the dominant conventional DC population of the spleen, but only a minor population in the thymus. Based on the expression of endothelial cell-specific adhesion molecule (ESAM), two distinct splenic CD11b+ populations can be distinguished [34]. ESAMhigh DC represent cells derived from DC progenitors while the ESAMlow population stems from circulating monocytes. In the periphery, CD8α−CD11b+ DC but not CD8α+ DC mediate cross-tolerance toward intestinal antigens [36]. In autoimmune diabetes, CD11b+ DC were recently shown to play a tolerogenic role [37]. A distinct tolerogenic subset of splenic CD11b+IDO+ DC from orally tolerized mice is responsible for induction of systemic immune tolerance and suppression of collagen-induced arthritis [38]. Antigen-induced tolerogenic CD11b+ DC are abundant in Peyer’s patches during the induction of oral tolerance to type II collagen and suppress experimental collagen-induced arthritis [39].
DC promote central tolerance
Unlike their established role as prototypic APCs initiating immune responses, the role of DC in steady state immune tolerance is less understood. Several studies have highlighted the role of DC in thymic T cell development. Targeted expression of major histocompatibility complex (MHC)-II molecules demonstrated that DC can induce negative but not positive selection of thymocytes in vivo [40] and hence mice with residual expression of MHC-II on mature DC fail to promote positive selection of CD4+ T cells [41]. Negative selection occurs when thymocytes are presented with and respond to self-antigens. Medullary thymic epithelial cells (mTEC) express many tissue-restricted self-antigens (TSA) in an autoimmune regulator (AIRE)-dependent manner [42]. Thymic DC can either directly present or cross-present self-antigens shed by mTEC. Thus, self-antigen presentation by thymic DC can either result in negative selection [43–45] or lead to the generation of thymus-derived Tregs (nTregs) [46]. As DC are highly migratory cells, thymic clonal deletion and/or nTreg development can also be driven by circulating DC presenting peripheral TSA [47]. Nevertheless, constitutive deletion of DC does not result in breakdown of central tolerance, as mice lacking DC exhibit a relatively normal T cell repertoire [48], indicating that DC are capable of, yet not mandatory for induction of central tolerance. Similarly, DC-deficient mice were found to contain normal nTreg numbers in the steady state without developing spontaneous autoimmunity [49]. However, a third study described a strong autoimmune lymphoproliferative infiltration followed by spontaneous fatal autoimmunity in mice that lack DC [50]. Our mice [48, 49] also show lymphoproliferative disease that is evident at around 4 months of age, but unlike the mice described by Ohnmacht et al. neither show signs of autoimmunity nor die from such disease. As both systems are genetically similar and depend on the same Cre-expressing mouse line, we have no good explanation for this discrepancy. One may speculate that differences in the gut microbiota are responsible for the generation of pathogenic responses in one mouse colony but not in the other, thereby contributing to development of fatal inflammatory bowel disease (IBD).
DC-mediated T cell homeostasis
Two photon imaging and live intravital microscopy had uncovered frequent and dynamic interactions between DC and T cells, even in the absence of infectious stimuli [51, 52]. T cell homeostasis and optimal functionality was found to be controlled by steady state DC, delivering tonic signals through MHC-T cell receptor (TCR) complex interactions [53, 54]. Similarly, Scheinecker et al. show that under steady state conditions, DC present self-antigen to T cells in the draining lymph nodes [29]. Thus, DC play a key role in maintaining immune homeostasis and promoting peripheral T cell tolerance, by continuously presenting self- or harmless foreign antigens to T cells in the absence of infection and inflammation, that is co-stimulation and/or activating cytokines [55, 56].
DC establish peripheral tolerance
Peripheral tolerance mechanisms necessitate from the limitations of central tolerance ‘avoiding horror autotoxicus’ [55]: (i) self-reactive lymphocytes escape negative selection, (ii) many innocuous environmental antigens, including commensal microbiota, are not expressed in the thymus and (iii) lymphocyte receptors for foreign antigens can cross-react with self. Early studies showing tolerance induction by DC used antigen-targeting in vivo by antibodies targeting receptors expressed specifically by DC [30, 57]. In this approach, one conjugates the self-antigen to a DC-specific antibody, for example anti-DEC205. Following injection of the antibody conjugate, it will bind to the specific receptor on DC, be internalized and the antigen will be processed and presented by the (immature) DC, which leads to tolerance in the absence of an adjuvants, e.g. TLR ligand or CD40L [56]. Despite this proof-of-concept, limitations of these early experiments using anti-DEC205–antigen conjugates to target DC were low-level expression of DEC205 and Fc receptor-mediated uptake by other cell types (macrophages, B cells). Thus, ongoing research aims to improve this strategy, for example, by identifying novel DC (subset)-specific endocytic receptors and generating better antibodies for improved DC-based immunotherapy of human disease.
By utilizing the genetic approach of Cre-recombinase, the group of Maries van den Broek demonstrated that induced expression of an immunodominant virus-derived antigen by DC results in strong CD8+ T cell unresponsiveness that could not be broken in wild type mice [58]. Follow-up studies shed light on the mechanism of DC-induced T cell tolerance, involving the expression of PD-1 and CTLA4 on CD8+ T cells and Treg induction [59, 60]. Using a similar approach, we have recently shown that induced expression of self-antigen by steady state DC induces robust CD4+ T cell tolerance [49]. Our data suggested that the mechanism instructing such tolerance involves the interaction of PD-L1 expressing DC with PD-1 expressing T cells, resulting in the generation of de novo antigen-specific peripheral iTregs. At the same time, constitutive DC-ablation results in enhanced autoimmunity following immunization with self-antigen [49].
In view of this tolerogenic role of DC, it was assumed that the loss of DC would result in a breakdown of peripheral tolerance. However, constitutive DC ablation did not lead to spontaneous autoimmunity but rather a myeloproliferative disorder [48]. Further studies identified an elevated Flt3L serum concentration as the driving force of this myeloproliferative syndrome due to the loss of DC [48, 61]. A similar scenario was reported in human patients with hereditary monocyte or DC deficiency [62]. Likewise, constitutive DC depletion in lupus prone MRL/lpr mice resulted in ameliorated autoimmunity. DC were shown to be crucial for the expansion and differentiation of T cells but were not required for their initial activation. In line with the above, kidney-interstitial infiltrates developed in the absence of DC, but failed to progress to an inflammatory disease [63]. In this model, systemic lupus erythematosus (SLE) becomes apparent at the age of 4 months. At this age, the mice are already partly immunodeficient (loss of tonic signals required for T cell homeostasis), harbouring reduced levels of peripheral T cells (both effector and regulatory) and to lesser extent B cells, which may explain the reduced autoimmunity. Taken together, these studies suggest that DC can induce peripheral T cell tolerance, yet they may not be essential for it. On the other hand, steady state DC appear to be mandatory for the generation of antigen-specific peripheral iTregs, by a mechanism that involves PD-L1/PD-1 interaction.
DC and Tregs
As indicated above, DC are important for maintaining peripheral T cell homeostasis and preventing inappropriate T cell activation. Extra-thymic or peripherally induced Treg cells (iTregs) are no exception. Several in vitro studies have shown that DC can induce induced Treg (iTreg) formation, particularly when Treg-promoting cytokines are added [3, 64, 65]. Yet, the absence of DC led only to a mild reduction of total Treg numbers at best [48, 66], although Treg homeostatic proliferation was shown to be dependent on the presence of DC [67]. Conversely, Treg ablation results in accelerated DC maturation and expansion [68], a process that was dependent on Flt3 [6]. In line with the above findings, several studies uncovered a direct correlation between DC and Treg numbers, as part of a feedback-control mechanism directed by Flt3L [66, 69]. In another report, Collins et al. identified a lamina propria CD103+ DC subset as the one subset that expands in response to Flt3L treatment [70]. CD103+ DC represent the tissue-resident DC subset equivalent to CD8+ DC in the lymph nodes and spleen, and Flt3L treatment skews DC toward the tolerogenic CD8+ lineage [71]. These studies suggest that there is a bidirectional feedback loop involving Flt3/Flt3L that regulates both CD8α+/CD103+ DC and Tregs. However, the development of CDP and subsequently the different DC progenies are also Flt3/Flt3L-dependent [1, 72], and Flt3L-dependent DC control immune responses to protein vaccine [73]. In addition, Flt3L might cause the expansion of other myeloid cells. Furthermore, the functionality of the DC that respond and expand in response to Flt3L might be different due to different pathways downstream of Flt3, e.g. PI3K, Akt and mTOR.
The tool kit of DC to generate Tregs
It is evident that DC support the differentiation and maintenance of different types of Treg cells, including the IL-10-producing Tr1, TGFβ-producing Th3, thymic-derived (nTregs) and peripherally induced (iTregs) Foxp3+ T cells. This is achieved by various mechanisms, including direct cell-cell contact-dependant signalling through surface molecules, as well as by affecting Treg cell fate through secretory proteins. These mechanisms are described in detail below and summarized in Fig. 2.

Dendritic cells mediate tolerance. a DC can mediate Treg generation via several surface molecules, including CD70, CD80/CD86, ICOS-L and PD-L1 or PD-L2. The expression of CD70 on DC induces a survival signal in thymic autoreactive Treg cells expressing CD27 and prevents them from undergoing negative selection. MHC-II antigen presentation by DC without an additional co-stimulatory signal (e.g. CD80/CD86), or in combination with a co-inhibitory signal (e.g. PD-L1/2) can lead to tolerance induction. Similarly, ligation of CD80/CD86 by cytotoxic T lymphocyte antigen 4 (CTLA4) drives Treg differentiation while insufficient ligation of CD80/CD86 by CD28 induces tolerance. The inducible T cell co-stimulator ligand (ICOS-L) expressed by DC binds to its receptor on T cells and maintains Treg homeostasis. b DC secrete many factors that are known to induce tolerance and Treg generation. Both TGFβ and IL-10 production by DC mediates Treg differentiation. At the same time, IL-10 also inhibits DC maturation, as well as MHC-II, co-stimulatory molecules and chemokine expression by DC. DC-derived IL-27 induces Treg generation by increasing IL-10 expression and repressing IL-1β and IL-23 production. DC are also an important source of retinoic acid (RA), which is involved in the generation of Tregs, while simultaneously inhibiting Th17 cells. Indoleamine 2,3-deoxygenase (IDO), giving rise to kynurenines and other tryptophan metabolites, is a strong inducer of T cell anergy, apoptosis and Treg differentiation. IDO may also induce TGFβ expression
Co-stimulatory signals delivered by cell-cell contact
CD80/86
Like all other T cell subtypes, Tregs also express CD28 and the interaction of CD28 with CD80/CD86 is required for normal thymic and peripheral Treg development and maintenance [74]. A recent study revealed that DC are an important source of CD80/86 to enhance Treg numbers [75]. Intriguingly, the CD80/86 signal delivered by DC was dispensable for the development of thymus-derived nTregs, but the loss of these molecules specifically on DC resulted in about 40 % reduction of peripherally induced iTregs.
CD70
CD70 is a TNF family member expressed on DC and mTECs (both AIRE+ and AIRE−), whereas its receptor CD27 is expressed on developing thymocytes. Recently, it has been discovered that the CD70–CD27 pathway plays an important role in thymic-derived nTreg development. Using CD70-deficient BM chimeric mice, Coquet et al. demonstrated that CD70-expressing CD8α+ DC (but not CD11b+ DC or pDC) contributed to nTreg development in the thymus. In particular, CD70–CD27 signalling drives the positive selection of nTregs and prevents them from undergoing apoptosis [76]. In the periphery, CD70 signalling promotes Th1 [77] and suppresses Th17 differentiation, which in turn results in reduced autoimmunity [78]. In contrast, in vitro experiments indicated that CD70-overexpression was not sufficient to drive Th17 differentiation, nor did it affect the generation of Tregs [79].
ICOS-L
In a model of airway inflammation, ICOS-L-expressing semi-mature DC promoted the induction of TGFβ-producing, antigen-specific iTregs [80]. Similarly, pDC stimulate the induction of iTregs in an ICOS-L-dependent manner [18]. In agreement, adoptive transfer of WT but not ICOS-L-deficient pDC protected mice against asthma [81].
PD-ligands
In contrast to WT APCs, when PD-L1-deficient APCs were used to generate iTregs in vitro, only a minimal iTreg conversion of naive CD4+ T cells was observed [82–84]. Recently, it has been reported that PD-1 ligation affects the stability of DC-T cell contacts [85]. Using a diabetes mouse model, T cells tolerized for self-antigen failed to form stable interactions with antigen-specific DC, but rather moved freely in the draining lymph nodes. Following administration of PD-L1 or PD-1 blocking antibodies, continued PD-L1 and PD-1 interactions inhibited TCR-mediated signal transduction [85]. Moreover, DC stimulation with soluble PD-1 suppresses DC maturation and promotes IL-10 secretion [86]. Recently, we have shown that upon transient or constitutive DC depletion, mice developed encephalomyelitis (EAE) after active immunization with the MOG peptide p35-55. Furthermore, PD-L1 expressing DC were crucial for in vivo generation of antigen-specific iTregs, which in turn dampens disease severity. De novo generation of antigen-specific iTregs was markedly elevated in cDC-proficient but not in cDC-deficient mice, whereas de novo generation of antigen-specific PD-1-deficient iTregs was comparable irrespective of the presence or absence of DC [49].
Signals conveyed by secreted molecules
Il-10
IL-10 is a regulatory cytokine produced by and acting on different immune cells, including T cells and DC [87–89]. While the effects of IL-10 on (regulatory) T cells are well established, its broad anti-inflammatory properties may also result from the ability to inhibit APC function, including their maturation, expression of MHC-II, co-stimulatory molecules, chemokines (both CC and CXC) and production of pro-inflammatory cytokines [90, 91]. IL-10 conditions human DC to acquire a tolerogenic phenotype favouring the induction of T cell anergy and suppressive function [92, 93]. Moreover, transfer of IL-10–treated DC limits effector T cell responses [94], protects mice from experimental autoimmune encephalomyelitis (EAE) and inhibits graft rejection in transplanted hosts [95, 96]. More recently, the in vivo role of IL-10 to restrain APC function is beginning to unfold. IL-10 controls DC in the skin to limit contact hypersensitivity and attenuate anti-Leishmania major immunity [97, 98]. In addition, IL-10 control of CD11c+ APCs and, in particular, CX3CR1+ macrophages is essential to maintain immune homeostasis in the intestine [99, 100]. Intriguingly, in the absence of IL-10 signalling in CD11c+ myeloid cells oral tolerance remains intact [100].
On the other hand, early in vitro studies have identified IL-10 production by DC to be an important mechanism in driving T cell anergy and suppression [101]. In vivo experiments had shown that following respiratory antigen challenge, IL-10 producing mature pulmonary DC induce tolerance via the generation of Tr1 cells [102]. BM-derived DC, differentiated in the presence of IL-10, GM-CSF and TNFα, developed into semi-mature DC, which following LPS-challenge produced high levels of IL-10 and promoted the differentiation of IL-10 producing suppressive T cells [103]. Similarly, Langerhans cells suppress contact hypersensitivity through IL-10 secretion, which in turn promotes Tr1 cell differentiation [104]. While IL-10–deficient mice develop spontaneous IBD by the age of 4–6 weeks [105], DC-specific deletion of IL-10 results in a mild spontaneous colitis, developing at a much older age (6–8 months) and with significantly lower incidence (Yogev and Waisman, unpublished observation). On the other hand, CX3CR1-specific and CD11c-specific deletion of IL-10 had no effect on, respectively, the development of colitis and EAE induction, severity or resolution [99] (Yogev and Waisman, unpublished observation).
Il-27
Co-culturing Tregs and DC results in the secretion of IL-10, IL-27 and TGFβ by DC, which further drives the differentiation of Tr1 cells [106]. DC-derived IL-27 suppresses the production of IL-1β and IL-23 and induces IL-10 secretion, thus blocking immunogenic Th17 differentiation and consequently autoimmunity [107]. At the same time, IL-27 drives the expression of c-Maf, IL-21 and ICOS in naïve T cells, which renders them to become Tr1 cells [108, 109]. DC-derived IL-27, via STAT-1 and STAT-3 activation, drives IL-10 transcription and activation of the IL-10 promoter, thus inducing Tr1 differentiation [109]. Importantly, it has been recently shown that the stimulation of human DC with IL-27 led to upregulation of PD-L1 surface expression, without leading to DC maturation [110]. In addition, it was shown recently that stimulation of DC with IL-27 might induce other regulatory pathways, such as the upregulation of CD39 and suppression of the inflammasome pathway [111].
TGFβ
Another well-known regulatory cytokine with pleiotropic effects on T cells as well as APCs is TGFβ [88, 90]. TGFβ promotes conversion of peripheral naive T cells into CD4+CD25+ Tregs by inducing Foxp3 expression [112–114]. Following LPS stimulation, a subset of splenic DC secretes high levels of TGFβ and drives Tr1 cell differentiation [115]. Similarly, several in vitro studies demonstrated that DC promote extra-thymic iTreg differentiation in a TGFβ-dependent manner [3, 64, 65, 92, 116, 117]. In vivo delivery of low-dose agonistic peptide to DC induces iTreg cell differentiation from antigen-specific naïve T cells. Inhibition of T cell-specific TGFβ signalling, by expression of a dominant-negative TGFβRII, blocked iTreg differentiation [32]. In line, CD11c-specific deletion of the TGFβRII leads to spontaneous multi-organ autoimmunity due to altered Treg cell function [118], although this phenotype may be partially mediated by TGFβ receptor-deficient T cells (Kel and Clausen, unpublished observation). The integrin α4β8 induces TGFβ activation via metalloproteinase-mediated degradation of latency-associated protein (LAP), resulting in the release of active TGFβ into the extracellular space [119]. DC-specific ablation of the α4β8 integrin results in activation and expansion of T cells, reduced levels of colonic Tregs and consequently the development of colitis [120]. In addition, TGFβ is required to maintain the pool of epidermal immature Langerhans cells, and hence, CD11c-specific TGFβRI-deficient mice develop reduced contact hypersensitivity due to the lack of these APCs [121].
Retinoic acid and β-catenin
DC-derived retinoic acid (RA) is a well-established mediator of oral tolerance induction via the generation of iTregs. Mucosal DC direct T cell homing into the gut, in a molecular mechanism that is attributed to DC-derived RA [122]. Moreover, Mucida et al. demonstrated that DC-derived RA has a reciprocal activity, inhibiting TGFβ-dependent Th17 cell generation while promoting Foxp3+ Treg cell differentiation [123]. Other studies revealed that RA promotes iTreg differentiation by blocking the generation of IFNγ- or IL-21–producing effector memory T cells [124]. Several publications reported that LP CD103+ DC induce peripheral Tregs by expressing aldehyde dehydrogenase (ALDH), an enzyme that metabolizes vitamin A into RA [125, 126]. Likewise, dermis-derived CD103− DC constitutively produce RA and induce Tregs [127].
A second signalling pathway that can induce RA-producing enzymes involves β-catenin, the central component of the canonical Wingless-Int (Wnt) signalling pathway [128], which is constitutively expressed in DC. Early studies had revealed a crucial role for β-catenin in the regulation of BM-DC maturation and function. In vitro disruption of E-cadherin/β-catenin binding in BM-DC results in their phenotypic maturation, i.e. upregulation of MHC-II and co-stimulatory molecules without activation of pro-inflammatory cytokine secretion, leading to a tolerogenic DC phenotype that promotes the induction of IL-10–producing Tregs [129]. In line, mice with a CD11c-specific deletion of β-catenin are more susceptible to DSS-induced colitis and EAE, which is accompanied by increased Th1/Th17 and reduced Foxp3+ Treg responses [130, 131]. Moreover, Wnt/β-catenin signalling contributes to tumour-induced immunosuppression by inhibiting DC cross-priming of CD8 T cells [132] via upregulation of IL-10 [133]. On the other hand, β-catenin signalling in DC supports the maintenance of CD8 T cells after clonal expansion [133] and drives the differentiation and pro-inflammatory function of IRF8-dependent DC subpopulations [134]. Our ongoing work indicates that mice with stabilized β-catenin in DC mount attenuated Th2 responses with reduced airway hyperresponsitivity and harbor more CD4+Foxp3+ Tregs after induction of allergic asthma (Ober-Blöbaum and Clausen, unpublished observation). Together, these observations led to the concept that β-catenin promotes a tolerogenic DC phenotype in vivo [135]. However, despite a lower frequency of Tregs, CD11c-specific β-catenin deficiency did not affect the severity or course of autoimmune collagen-induced arthritis [136].
Indoleamine-2,3-dioxygenase
Indoleamine-2,3-dioxygenase (IDO) catabolizes the essential amino acid tryptophan into the stable metabolite kynurenine. The latter results in cell starvation due to physical depletion of tryptophan from the local environment and activation of the general control nonderepressible 2 (GCN2) kinase that phosphorylates the eukaryotic initiation factor 2 (eIF2) (which activates the amino acid stress response) pathway that promotes Treg generation and expansion in an IDO-rich environment [137]. Furthermore, kynurenine can directly generate an immunosuppressive environment by binding and interacting with aryl hydrocarbon receptor (AhR) on CD4+ T cells, thus promoting their polarization into Tregs [138, 139]. In addition, AhR can also upregulate the expression of IDO and RA-producing enzymes directly in the DC [139, 140].
DC production of IDO plays a critical role in tolerance induction by promoting iTreg generation [141]. In turn, iTregs can induce the production of IDO through reverse signalling in pDC [142]. IDO production differs between different DC subsets, with CD8α+ DC producing higher levels of IDO compared to CD8α– DC [143]. Intestinal CD103+ DC also produce high levels of IDO and play a central role in maintaining gut homeostasis and oral tolerance [144]. Furthermore, ligation of CD80/86 on DC with CTLA4 induces IDO expression [145–147]. IDO-deficient mice developed exacerbated EAE, which could be prevented by treatment with the tryptophan metabolite 3-HAA. 3-HAA instructs DC to produce TGFβ and blocks IL-6 synthesis, thus promoting Treg and at the same time inhibiting Th17 differentiation [148].
Outlook: treatment of autoimmune diseases with DC
DC hold a great promise for the therapy of human diseases. On the one hand, DC may enhance anti-tumour immunity when attempting to fight cancer. On the other hand, they may induce tolerance, which is essential in case of transplantation and autoimmunity. Nonetheless, here lies their main danger: the potential threat that the transferred cells may change once within the patients, and thus cause tolerance instead of immunity, and vice versa.
In recent years, much effort was put into establishing protocols to induce tolerogenic DC and retain them as such also after transfer [149, 150]. To obtain tolerogenic DC, one can either culture them using mixtures of cytokines and stimuli that results in their generation, or genetically manipulate DC so that they express less immunogenic molecules, leading to DC that induce tolerance rather than immunity. As indicated above, deletion of co-stimulatory molecules, e.g. deficiency of B7-H1 (PD-L1), lead to the generation of tolerogenic DC in the mouse system [151]. But obviously, this is not possible to achieve in humans, so other protocols were established by using cytokine cocktails, including for example IL-10 or TNFα [152–154]. These protocols led to the generation of DC that were able to induce profound tolerance in mouse models of different autoimmune diseases, including graft-versus-host disease [155], collagen-induced arthritis [156], autoimmune thyroiditis [157] and EAE [154].
In humans, it is obviously not possible to generate DC from genetically manipulated individuals; however, it is possible to manipulate human DC gene expression using siRNA. Using such methods, it is possible to block the expression of co-stimulatory molecules or enhance the expression of IDO or TGFβ by DC (for review see [150]). One can also generate, similarly to what was done before in the mouse system, tolerogenic DC from the patients using cytokine cocktails. Such protocols were rather successful in initial experiments [158–160]. Once tolerogenic DC are established, they must be put to test in clinical trials. There were not many clinical trials performed to date with DC, and most of the ones that were performed were done with the aim to boost immunity in cancer therapy and not using tolerogenic DC to treat autoimmunity. A comprehensive review of the up-to-date clinical trials was written by Galluzzi et al. [161, 162]. It is evident that although many phase I and a few phase II experiments were initiated, these trials were found to be safe for the patients, but on the other hand, no clear advantage was reported so far. More trials should be performed to establish beneficial effects for the patients. The lack of side effects in these trials is therefore helpful also in the new attempts to treat autoimmune diseases with DC, such as the trial reported by Giannoukakis et al. for type I diabetes [45, 163].
References
Geissmann F et al. (2010) Development of monocytes, macrophages, and dendritic cells. Science 327(5966):656–661
Naik SH et al. (2007) Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol 8(11):1217–1226
Sela U et al. (2011) Dendritic cells induce antigen-specific regulatory T cells that prevent graft versus host disease and persist in mice. J Exp Med 208(12):2489–2496
Bogunovic M et al. (2009) Origin of the lamina propria dendritic cell network. Immunity 31(3):513–525
Ginhoux F et al. (2009) The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med 206(13):3115–3130
Liu K et al. (2009) In vivo analysis of dendritic cell development and homeostasis. Science 324(5925):392–397
Merad M et al. (2013) The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 31:563–604
Colonna M, Trinchieri G, Liu YJ (2004) Plasmacytoid dendritic cells in immunity. Nat Immunol 5(12):1219–1226
Reizis B et al. (2011) Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol 29:163–183
Nakano H, Yanagita M, Gunn MD (2001) CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J Exp Med 194(8):1171–1178
Asselin-Paturel C et al. (2001) Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol 2(12):1144–1150
Bjorck P (2001) Isolation and characterization of plasmacytoid dendritic cells from Flt3 ligand and granulocyte-macrophage colony-stimulating factor-treated mice. Blood 98(13):3520–3526
Kuwana M (2002) Induction of anergic and regulatory T cells by plasmacytoid dendritic cells and other dendritic cell subsets. Hum Immunol 63(12):1156–1163
Moseman EA et al. (2004) Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4 + CD25+ regulatory T cells. J Immunol 173(7):4433–4442
Boonstra A et al. (2003) Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll-like receptor ligation. J Exp Med 197(1):101–109
Martin P et al. (2002) Characterization of a new subpopulation of mouse CD8alpha + B220+ dendritic cells endowed with type 1 interferon production capacity and tolerogenic potential. Blood 100(2):383–390
Bilsborough J et al. (2003) Mucosal CD8alpha + DC, with a plasmacytoid phenotype, induce differentiation and support function of T cells with regulatory properties. Immunology 108(4):481–492
Ito T et al. (2007) Plasmacytoid dendritic cells prime IL-10-producing T regulatory cells by inducible costimulator ligand. J Exp Med 204(1):105–115
McKenna HJ et al. (2000) Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood 95(11):3489–3497
Waskow C et al. (2008) The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol 9(6):676–683
Hacker C et al. (2003) Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol 4(4):380–386
Kusunoki T et al. (2003) TH2 dominance and defective development of a CD8+ dendritic cell subset in Id2-deficient mice. J Allergy Clin Immunol 111(1):136–142
Holtschke T et al. (1996) Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell 87(2):307–317
Schiavoni G et al. (2002) ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med 196(11):1415–1425
Hildner K et al. (2008) Batf3 deficiency reveals a critical role for CD8alpha + dendritic cells in cytotoxic T cell immunity. Science 322(5904):1097–1100
Edelson BT et al. (2010) Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha + conventional dendritic cells. J Exp Med 207(4):823–836
Yamazaki S et al. (2008) CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J Immunol 181(10):6923–6933
Belz GT et al. (2002) The CD8alpha(+) dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. J Exp Med 196(8):1099–1104
Scheinecker C et al. (2002) Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J Exp Med 196(8):1079–1090
Hawiger D et al. (2001) Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med 194(6):769–779
Bonifaz L et al. (2002) Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med 196(12):1627–1638
Kretschmer K et al. (2005) Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol 6(12):1219–1227
Fukaya T et al. (2012) Conditional ablation of CD205+ conventional dendritic cells impacts the regulation of T-cell immunity and homeostasis in vivo. Proc Natl Acad Sci U S A 109(28):11288–11293
Lewis KL et al. (2011) Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity 35(5):780–791
Greter M et al. (2012) GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity 36(6):1031–1046
Chung Y et al. (2005) CD8alpha-11b + dendritic cells but not CD8alpha + dendritic cells mediate cross-tolerance toward intestinal antigens. Blood 106(1):201–206
Kriegel MA, Rathinam C, Flavell RA (2012) Pancreatic islet expression of chemokine CCL2 suppresses autoimmune diabetes via tolerogenic CD11c + CD11b + dendritic cells. Proc Natl Acad Sci U S A 109(9):3457–3462
Park MJ et al. (2012) A distinct tolerogenic subset of splenic IDO(+)CD11b(+) dendritic cells from orally tolerized mice is responsible for induction of systemic immune tolerance and suppression of collagen-induced arthritis. Cell Immunol 278(1–2):45–54
Min SY et al. (2006) Antigen-induced, tolerogenic CD11c+,CD11b + dendritic cells are abundant in Peyer’s patches during the induction of oral tolerance to type II collagen and suppress experimental collagen-induced arthritis. Arthritis Rheum 54(3):887–898
Brocker T, Riedinger M, Karjalainen K (1997) Targeted expression of major histocompatibility complex (MHC) class II molecules demonstrates that dendritic cells can induce negative but not positive selection of thymocytes in vivo. J Exp Med 185(3):541–550
Clausen BE et al. (1998) Residual MHC class II expression on mature dendritic cells and activated B cells in RFX5-deficient mice. Immunity 8(2):143–155
Peterson P, Org T, Rebane A (2008) Transcriptional regulation by AIRE: molecular mechanisms of central tolerance. Nat Rev Immunol 8(12):948–957
Hubert FX et al. (2011) Aire regulates the transfer of antigen from mTECs to dendritic cells for induction of thymic tolerance. Blood 118(9):2462–2472
Klein L et al. (2011) Autonomous versus dendritic cell-dependent contributions of medullary thymic epithelial cells to central tolerance. Trends Immunol 32(5):188–193
Aichinger M et al. (2013) Macroautophagy substrates are loaded onto MHC class II of medullary thymic epithelial cells for central tolerance. J Exp Med 210(2):287–300
Lei Y et al. (2011) Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory T cell development. J Exp Med 208(2):383–394
Proietto AI, van Dommelen S, Wu L (2009) The impact of circulating dendritic cells on the development and differentiation of thymocytes. Immunol Cell Biol 87(1):39–45
Birnberg T et al. (2008) Lack of conventional dendritic cells is compatible with normal development and T cell homeostasis, but causes myeloid proliferative syndrome. Immunity 29(6):986–997
Yogev N et al. (2012) Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor(+) regulatory T cells. Immunity 37(2):264–275
Ohnmacht C et al. (2009) Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med 206(3):549–559
Stoll S et al. (2002) Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science 296(5574):1873–1876
Shakhar G et al. (2005) Stable T cell-dendritic cell interactions precede the development of both tolerance and immunity in vivo. Nat Immunol 6(7):707–714
Hochweller K et al. (2010) Dendritic cells control T cell tonic signaling required for responsiveness to foreign antigen. Proc Natl Acad Sci U S A 107(13):5931–5936
Garbi N et al. (2010) Tonic T cell signalling and T cell tolerance as opposite effects of self-recognition on dendritic cells. Curr Opin Immunol 22(5):601–608
Steinman RM, Nussenzweig MC (2002) Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A 99(1):351–358
Steinman RM, Hawiger D, Nussenzweig MC (2003) Tolerogenic dendritic cells. Annu Rev Immunol 21:685–711
Dudziak D et al. (2007) Differential antigen processing by dendritic cell subsets in vivo. Science 315(5808):107–111
Probst HC et al. (2003) Inducible transgenic mice reveal resting dendritic cells as potent inducers of CD8+ T cell tolerance. Immunity 18(5):713–720
Probst HC et al. (2005) Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol 6(3):280–286
Schildknecht A et al. (2010) FoxP3+ regulatory T cells essentially contribute to peripheral CD8+ T-cell tolerance induced by steady-state dendritic cells. Proc Natl Acad Sci U S A 107(1):199–203
Hochweller K et al. (2009) Homeostasis of dendritic cells in lymphoid organs is controlled by regulation of their precursors via a feedback loop. Blood 114(20):4411–4421
Collin M et al. (2011) Human dendritic cell deficiency: the missing ID? Nat Rev Immunol 11(9):575–583
Teichmann LL et al. (2010) Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity 33(6):967–978
Yamazaki S et al. (2003) Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J Exp Med 198(2):235–247
Tarbell KV et al. (2004) CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med 199(11):1467–1477
Darrasse-Jeze G et al. (2009) Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med 206(9):1853–1862
Suffner J et al. (2010) Dendritic cells support homeostatic expansion of Foxp3+ regulatory T cells in Foxp3.LuciDTR mice. J Immunol 184(4):1810–1820
Kim JM, Rasmussen JP, Rudensky AY (2007) Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 8(2):191–197
Swee LK et al. (2009) Expansion of peripheral naturally occurring T regulatory cells by fms-like tyrosine kinase 3 ligand treatment. Blood 113(25):6277–6287
Collins CB et al. (2012) Flt3 ligand expands CD103(+) dendritic cells and FoxP3(+) T regulatory cells, and attenuates Crohn’s-like murine ileitis. Gut 61(8):1154–1162
Vollstedt S et al. (2004) Treatment of neonatal mice with Flt3 ligand leads to changes in dendritic cell subpopulations associated with enhanced IL-12 and IFN-alpha production. Eur J Immunol 34(7):1849–1860
Belz GT, Nutt SL (2012) Transcriptional programming of the dendritic cell network. Nat Rev Immunol 12(2):101–113
Anandasabapathy N et al. (2014) Classical Flt3L-dependent dendritic cells control immunity to protein vaccine. J Exp Med 211(9):1875–1891
Salomon B et al. (2000) B7/CD28 costimulation is essential for the homeostasis of the CD4 + CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12(4):431–440
Bar-On L et al. (2011) Dendritic cell-restricted CD80/86 deficiency results in peripheral regulatory T-cell reduction but is not associated with lymphocyte hyperactivation. Eur J Immunol 41(2):291–298
Coquet JM et al. (2013) Epithelial and dendritic cells in the thymic medulla promote CD4 + Foxp3+ regulatory T cell development via the CD27-CD70 pathway. J Exp Med 210(4):715–728
Soares H et al. (2007) A subset of dendritic cells induces CD4+ T cells to produce IFN-gamma by an IL-12-independent but CD70-dependent mechanism in vivo. J Exp Med 204(5):1095–1106
Coquet JM et al. (2013) The CD27 and CD70 costimulatory pathway inhibits effector function of T helper 17 cells and attenuates associated autoimmunity. Immunity 38(1):53–65
Libregts S et al. (2011) Function of CD27 in helper T cell differentiation. Immunol Lett 136(2):177–186
Akbari O et al. (2002) Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat Med 8(9):1024–1032
Kool M et al. (2009) An anti-inflammatory role for plasmacytoid dendritic cells in allergic airway inflammation. J Immunol 183(2):1074–1082
Wang C et al. (2010) Down-modulation of programmed death 1 alters regulatory T cells and promotes experimental autoimmune encephalomyelitis. J Neurosci Res 88(1):7–15
Francisco LM et al. (2009) PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 206(13):3015–3029
Wang L et al. (2008) Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3 + CD4+ regulatory T cells. Proc Natl Acad Sci U S A 105(27):9331–9336
Fife BT et al. (2009) Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol 10(11):1185–1192
Kuipers H et al. (2006) Contribution of the PD-1 ligands/PD-1 signaling pathway to dendritic cell-mediated CD4+ T cell activation. Eur J Immunol 36(9):2472–2482
Moore KW et al. (2001) Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 19:683–765
Li MO, Flavell RA (2008) Contextual regulation of inflammation: a duet by transforming growth factor-beta and interleukin-10. Immunity 28(4):468–476
Saraiva M, O’Garra A (2010) The regulation of IL-10 production by immune cells. Nat Rev Immunol 10(3):170–181
Clausen BE, Kel JM (2010) Langerhans cells: critical regulators of skin immunity? Immunol Cell Biol 88(4):351–360
Clausen BE, Girard-Madoux MJ (2013) IL-10 control of dendritic cells in the skin. Oncoimmunology 2(3):e23186
Torres-Aguilar H et al. (2010) Tolerogenic dendritic cells generated with different immunosuppressive cytokines induce antigen-specific anergy and regulatory properties in memory CD4+ T cells. J Immunol 184(4):1765–1775
Steinbrink K et al. (1999) Interleukin-10-treated human dendritic cells induce a melanoma-antigen-specific anergy in CD8(+) T cells resulting in a failure to lyse tumor cells. Blood 93(5):1634–1642
Muller G et al. (2002) Interleukin-10-treated dendritic cells modulate immune responses of naive and sensitized T cells in vivo. J Investig Dermatol 119(4):836–841
Perona-Wright G et al. (2007) IL-10 permits transient activation of dendritic cells to tolerize T cells and protect from central nervous system autoimmune disease. Int Immunol 19(9):1123–1134
Lan YY et al. (2006) “Alternatively activated” dendritic cells preferentially secrete IL-10, expand Foxp3 + CD4+ T cells, and induce long-term organ allograft survival in combination with CTLA4-Ig. J Immunol 177(9):5868–5877
Girard-Madoux MJ et al. (2012) IL-10 controls dendritic cell-induced T-cell reactivation in the skin to limit contact hypersensitivity. J Allergy Clin Immunol 129(1):143–150 e1-10
Girard-Madoux MJ et al. (2015) IL-10 signaling in dendritic cells attenuates anti-Leishmania major immunity without affecting protective memory responses. J Investig Dermatol 135(11):2890–2894
Zigmond E et al. (2014) Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity 40(5):720–733
Girard-Madoux MJ et al. (2016) IL-10 control of CD11c + myeloid cells is essential to maintain immune homeostasis in the small and large intestine. Oncotarget. doi:10.18632/oncotarget.8337
Jonuleit H et al. (2000) Induction of interleukin 10-producing, nonproliferating CD4(+) T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med 192(9):1213–1222
Akbari O, DeKruyff RH, Umetsu DT (2001) Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat Immunol 2(8):725–731
Wakkach A et al. (2003) Characterization of dendritic cells that induce tolerance and T regulatory 1 cell differentiation in vivo. Immunity 18(5):605–617
Igyarto BZ et al. (2009) Langerhans cells suppress contact hypersensitivity responses via cognate CD4 interaction and langerhans cell-derived IL-10. J Immunol 183(8):5085–5093
Kuhn R et al. (1993) Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75(2):263–274
Awasthi A et al. (2007) A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol 8(12):1380–1389
Sweeney CM et al. (2011) IL-27 mediates the response to IFN-beta therapy in multiple sclerosis patients by inhibiting Th17 cells. Brain Behav Immun 25(6):1170–1181
Pot C et al. (2009) Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J Immunol 183(2):797–801
Wang H et al. (2011) IL-27 induces the differentiation of Tr1-like cells from human naive CD4+ T cells via the phosphorylation of STAT1 and STAT3. Immunol Lett 136(1):21–28
Karakhanova S et al. (2011) IL-27 renders DC immunosuppressive by induction of B7-H1. J Leukoc Biol 89(6):837–845
Mascanfroni ID et al. (2013) IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol 14(10):1054–1063
Chen W et al. (2003) Conversion of peripheral CD4 + CD25- naive T cells to CD4 + CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 198(12):1875–1886
Marie JC, Liggitt D, Rudensky AY (2006) Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity 25(3):441–454
Gorelik L, Flavell RA (2000) Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 12(2):171–181
Zhang X et al. (2005) CD4-8- dendritic cells prime CD4+ T regulatory 1 cells to suppress antitumor immunity. J Immunol 175(5):2931–2937
Geissmann F et al. (1999) TGF-beta 1 prevents the noncognate maturation of human dendritic Langerhans cells. J Immunol 162(8):4567–4575
Ohtani T et al. (2009) TGF-beta1 dampens the susceptibility of dendritic cells to environmental stimulation, leading to the requirement for danger signals for activation. Immunology 126(4):485–499
Ramalingam R et al. (2012) Dendritic cell-specific disruption of TGF-beta receptor II leads to altered regulatory T cell phenotype and spontaneous multiorgan autoimmunity. J Immunol 189(8):3878–3893
Mu D et al. (2002) The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol 157(3):493–507
Travis MA et al. (2007) Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature 449(7160):361–365
Kel JM et al. (2010) TGF-beta is required to maintain the pool of immature Langerhans cells in the epidermis. J Immunol 185(6):3248–3255
Iwata M et al. (2004) Retinoic acid imprints gut-homing specificity on T cells. Immunity 21(4):527–538
Mucida D et al. (2007) Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317(5835):256–260
Hill JA et al. (2008) Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4 + CD44hi cells. Immunity 29(5):758–770
Sun CM et al. (2007) Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 204(8):1775–1785
Coombes JL et al. (2007) A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 204(8):1757–1764
Guilliams M et al. (2010) Skin-draining lymph nodes contain dermis-derived CD103(−) dendritic cells that constitutively produce retinoic acid and induce Foxp3(+) regulatory T cells. Blood 115(10):1958–1968
Staal FJ, Luis TC, Tiemessen MM (2008) WNT signalling in the immune system: WNT is spreading its wings. Nat Rev Immunol 8(8):581–593
Jiang A et al. (2007) Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity 27(4):610–624
Manicassamy S et al. (2010) Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 329(5993):849–853
Suryawanshi A et al. (2015) Canonical wnt signaling in dendritic cells regulates Th1/Th17 responses and suppresses autoimmune neuroinflammation. J Immunol 194(7):3295–3304
Liang X et al. (2014) Beta-catenin mediates tumor-induced immunosuppression by inhibiting cross-priming of CD8(+) T cells. J Leukoc Biol 95(1):179–190
Fu C et al. (2015) Beta-catenin in dendritic cells exerts opposite functions in cross-priming and maintenance of CD8+ T cells through regulation of IL-10. Proc Natl Acad Sci U S A 112(9):2823–2828
Cohen SB et al. (2015) Beta-catenin signaling drives differentiation and proinflammatory function of IRF8-dependent dendritic cells. J Immunol 194(1):210–222
Mellman I, Clausen BE (2010) Immunology. Beta-catenin balances immunity. Science 329(5993):767–769
Alves CH et al. (2015) Dendritic cell-specific deletion of beta-catenin results in fewer regulatory T-cells without exacerbating autoimmune collagen-induced arthritis. PLoS One 10(11):e0142972
Fallarino F et al. (2006) The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol 176(11):6752–6761
Mezrich JD et al. (2010) An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 185(6):3190–3198
Nguyen NT et al. (2010) Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A 107(46):19961–19966
Quintana FJ et al. (2010) An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 107(48):20768–20773
Munn DH, Mellor AL (2007) Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest 117(5):1147–1154
Grohmann U et al. (2007) Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med 13(5):579–586
Fallarino F et al. (2002) Functional expression of indoleamine 2,3-dioxygenase by murine CD8 alpha(+) dendritic cells. Int Immunol 14(1):65–68
Matteoli G et al. (2010) Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut 59(5):595–604
Grohmann U et al. (2002) CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol 3(11):1097–1101
Fallarino F et al. (2003) Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol 4(12):1206–1212
Mellor AL et al. (2004) Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol 16(10):1391–1401
Yan Y et al. (2010) IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol 185(10):5953–5961
Lutz MB (2012) Therapeutic potential of semi-mature dendritic cells for tolerance induction. Front Immunol 3:123
Hilkens CM, Isaacs JD, Thomson AW (2010) Development of dendritic cell-based immunotherapy for autoimmunity. Int Rev Immunol 29(2):156–183
Brandl C et al. (2010) B7-H1-deficiency enhances the potential of tolerogenic dendritic cells by activating CD1d-restricted type II NKT cells. PLoS One 5(5):e10800
Kleindienst P et al. (2005) Simultaneous induction of CD4 T cell tolerance and CD8 T cell immunity by semimature dendritic cells. J Immunol 174(7):3941–3947
Lim DS et al. (2009) Semi-mature DC are immunogenic and not tolerogenic when inoculated at a high dose in collagen-induced arthritis mice. Eur J Immunol 39(5):1334–1343
Menges M et al. (2002) Repetitive injections of dendritic cells matured with tumor necrosis factor alpha induce antigen-specific protection of mice from autoimmunity. J Exp Med 195(1):15–21
Sato K et al. (2003) Regulatory dendritic cells protect mice from murine acute graft-versus-host disease and leukemia relapse. Immunity 18(3):367–379
Stoop JN et al. (2010) Therapeutic effect of tolerogenic dendritic cells in established collagen-induced arthritis is associated with a reduction in Th17 responses. Arthritis Rheum 62(12):3656–3665
Verginis P, Li HS, Carayanniotis G (2005) Tolerogenic semimature dendritic cells suppress experimental autoimmune thyroiditis by activation of thyroglobulin-specific CD4 + CD25+ T cells. J Immunol 174(11):7433–7439
Dhodapkar MV et al. (2001) Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med 193(2):233–238
Fong L et al. (2001) Dendritic cells injected via different routes induce immunity in cancer patients. J Immunol 166(6):4254–4259
Boks MA et al. (2012) IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction—a comparative study of human clinical-applicable DC. Clin Immunol 142(3):332–342
Galluzzi L et al. (2012) Trial watch: dendritic cell-based interventions for cancer therapy. Oncoimmunology 1(7):1111–1134
Kepp O et al. (2012) Anticancer activity of cardiac glycosides: at the frontier between cell-autonomous and immunological effects. Oncoimmunology 1(9):1640–1642
Giannoukakis N et al. (2011) Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 34(9):2026–2032
Acknowledgments
We thank Tommy Regen, Ronald Backer and Julia Ober-Blöbaum for critical reading of the manuscript. Research that is relevant for this review is and has been supported by grants from, respectively, the DFG (SFB TR128 and TR156 to AW) and the NWO (VIDI 917-76-365 to BEC) as well as by the Forschungszentrum für Immuntherapie (FZI) Mainz.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
This article is a contribution to the special issue on Dendritic Cell Subsets and Immune-mediated Diseases - Guest Editor: Francisco Quintana
Rights and permissions
About this article

Cite this article
Waisman, A., Lukas, D., Clausen, B.E. et al. Dendritic cells as gatekeepers of tolerance. Semin Immunopathol 39, 153–163 (2017). https://doi.org/10.1007/s00281-016-0583-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-016-0583-z