
Abstract
The recruitment of specific leukocyte subtypes to the site of tissue injury is the cornerstone of inflammation and disease progression. This process has become an intense area of research because it presents several possible steps against which disease-specific therapies could be targeted. Leukocytes are recruited out of the blood stream by a series of events that include their capture, rolling, activation, and migration along the endothelium. In the last step, the leukocytes squeeze between adjacent endothelial cells to gain access to the inflamed tissue through a process referred to as transendothelial migration (TEM). Although many of the molecules, such as PECAM and CD99, that regulate these sequential steps have been identified, much less is understood regarding how they work together to coordinate the complex intercellular communications and dramatic shape changes that take place between the endothelial cells and leukocytes. Several of the endothelial cell proteins that function in TEM are localized to the lateral border recycling compartment (LBRC), an interconnected reticulum of membrane that recycles selectively to the endothelial borders. The recruitment of the LBRC to surround the migrating leukocyte is required for efficient TEM. This review will focus on the proteins and mechanisms that mediate TEM and specifically how the LBRC functions in the context of these molecular interactions and membrane movements.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
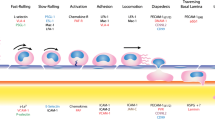
Inflammation is the body’s attempt to “right the ship” in response to tissue damage caused by virtually any inciting agent. Although the leukocytes subset(s) that are mobilized into action depends heavily on the type and location of the insult, all leukocytes must cross the endothelial barrier to enter tissue and execute their function. The overall process of leukocyte exit from the bloodstream, referred to as extravasation, involves a complex series of molecular and signaling events (reviewed in [1–3] and other chapters in this volume). Cytokines and other proinflammatory mediators secreted from the inflamed tissue trigger changes in the local endothelium that produce both an immediate increase in the expression of P-selectin and the de novo synthesis of E-selectin as well as increases in the synthesis and expression of ICAM-1 and VCAM-1. Selectin ligands on passing leukocytes can then interact with the selectins to slow the cell and bring it out of the flow of the bloodstream. The captured leukocyte then rolls along the endothelium until stimulation of its chemokine receptors by endothelial surface-bound chemokines or other mediators induces inside-out signaling events. These shift the leukocyte integrins to a “high-affinity” state that allows the rolling leukocyte to arrest and adhere tightly via integrin binding to ICAM-1, ICAM-2, and/or VCAM-1. The bound leukocyte then crawls along the surface of the endothelium, largely by Mac1/ICAM-1 interactions until it reaches an appropriate endothelial border, across which it migrates in the process of diapedesis. The early steps in leukocyte extravasation are relatively well characterized and have been reviewed elsewhere [4–6], and will not be discussed further here. Instead, this review will focus on the latter steps in extravasation with particular focus on the molecules and mechanisms involved in process of diapedesis, in which the leukocyte squeezes through the endothelium.
The process of transendothelial migration
Endothelial cells
The lumen of all blood vessels is lined by endothelial cells, which, in essence, bridge the tissue/fluid interface and represent the first barrier faced by leukocytes as they exit the bloodstream. The vascular endothelium allows for passage of solutes but must prevent the leakage of serum components and exposure of the basement membrane to clotting factors in the blood. As such, the typical endothelium is permeable to small molecules but acts as sieve preventing platelets, red blood cells, and leukocytes from passively diffusing through. The thin endothelial cells (<1 μm thick at the periphery) facilitate this selective permeability by regulating both the transport of solute across their cell body and their interactions with adjacent endothelial cells [7]. Although endothelial cells all perform common functions regardless of the vessel size or location, there are notable differences that have been reported in different vascular beds. For example, endothelial cells at the blood brain barrier (BBB) have significantly stronger inter-endothelial cell adhesions which are thought to function in limiting and regulating the passage of potentially deleterious molecules and inflammatory cells into the central nervous system (more on this specialized endothelium below) [8, 9]. Differences in endothelial morphology are also observed in various vascular beds although it is not clear if the morphological differences in the endothelial cells are due to distinct gene expression profiles and biochemical constitutions or inherent differences in blood velocity and volume of different vessels [10, 11]. Indeed, cultured endothelial cells are able to sense flow and respond by both elongating in the direction of the flow and activating various signaling pathways (reviewed in [12–14]). Conversely, endothelial cells also interact with and respond to their microenvironment suggesting that they receive important cues from the surrounding cells and tissue [11]. This is perhaps most apparent at the BBB where endothelial cells closely interact with astrocytes. In vitro treatment of endothelial cells from peripheral tissues with media collected from astrocytes is sufficient to induce a more “BBB-like’” phenotype suggesting that some soluble factors secreted by astrocytes can influence endothelial cell function [15, 16].
Transendothelial migration
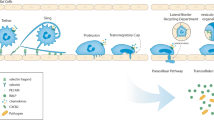
Transendothelial migration (TEM, also known as diapedesis) typically occurs at the borders of endothelial cells (paracellular TEM) [17, 18]. Leukocytes can also migrate through the endothelial cell body (transcellular TEM), although under most conditions this occurs in less than 10 % of events in vitro and in vivo [19, 20]. TEM is thought to be the committed step in the process as leukocytes at each of the earlier steps of capture, rolling, adhesion, and locomotion can detach and re-enter the circulation whereas leukocytes that have undergone TEM rarely if ever reverses the process [21]. As such, TEM is an excellent target for the design of therapeutic interventions. This is made even more attractive by the possibility of differences in the various vascular beds which could, in theory, could be exploited to regulate inflammation in a location or organ specific manner. Most leukocyte TEM occurs in post-capillary venules, although the exact reasons for this are largely unknown.
VE-cadherin and adherens junctions
Because most TEM occurs at the endothelial cell borders (paracellular), the proteins, and mechanisms that regulate these intercellular contacts has received significant attention. Inter-endothelial cell adhesion in post-capillary venules is primarily facilitated by adherens junctions, which are present around the entire lateral membrane of the cell [22, 23]. A main component of adherens junctions is vascular endothelial cadherin (VE-cadherin, cadherin-5), a member of the classical cadherin superfamily. VE-cadherin is expressed as a ∼125 kDa Type 1 membrane glycoprotein and contains 5 extracellular (EC) domains, of which the N-terminal EC domain 1 facilitates homophilic interactions with EC domain 1, and possibly domains 4 and 5, of VE-cadherin on an adjacent cell [24]. The intracellular tail of VE-cadherin has been shown to interact with p120, β-catenin, and plakoglobin [25]. The complex is linked to the actin cytoskeleton through α-catenin, which interacts with both β-catenin and plakoglobin [26]. This macromolecular association is critical to stabilizing the junction and maintaining the selective permeability and barrier function of the endothelium. Indeed, virtually all treatments and mechanisms that increase vascular permeability appear to disrupt these interactions. Common to the mechanisms that enhance vascular permeability in response to diverse stimuli is the phosphorylation of the VE-cadherin cytoplasmic tail. This phosphorylation disrupts its interaction with p120 and β-catenin, thus untethering it from the actin cytoskeleton [25–28]. The phosphorylation events and related signaling mechanisms have been reviewed extensively elsewhere [24]. As most transendothelial migration occurs at cell–cell contacts and adherens junctions are present around the lateral membrane of the cell, the migrating leukocyte must contend with this barrier in order to gain access to the tissue beneath.
VE-cadherin function also appears to be regulated by its association with vascular endothelial protein tyrosine phosphatase (VE-PTP) [29, 30]. Under resting conditions, VE-PTP interacts with VE-cadherin to stabilize it at cell–cell contacts and maintain junctional integrity, possibly by keeping it in a hypo-phosphorylated state [31]. Vestweber and coworkers have elegantly shown that this complex is dissociated in vivo during LPS-induced inflammation or upon stimulation of by VEGF. Furthermore, this dissociation is an essential step in opening the junctions for both permeability increases and leukocyte extravasation [29, 31]. These findings add another layer of regulation to the ever evolving model of VE-cadherin phosphorylation during junctional remodeling and TEM.
It is important to point out that, while adherens junctions play a role in maintaining vascular permeability and as a barrier to leukocyte egress, and while enhanced junctional permeability and increased leukocyte emigration are both part of the inflammatory response, they are distinct and separable events. The peak of vascular permeability occurs prior to the onset of leukocyte emigration. Furthermore, these can now be distinguished mechanistically, since mice deficient in cortactin (to be discussed in detail below) have inherently increased permeability but actually show a deficiency in TEM [32].
Tight junctions
In addition to adherens junctions, endothelial cells at the BBB also have tight junctions. Like adherens junctions, tight junctions are facilitated through transmembrane proteins (especially Claudins 1 and 5 and Occludin) that interact with each other across the junction and link to the actin cytoskeleton (through ZO-1) around the entire periphery of the endothelial cell. As the name suggests, tight junctions form a significantly stronger barrier that is reported to have greater than tenfold reduced permeability compared to adherens junctions [8, 9]. Transendothelial migration of leukocytes across the BBB does occur, although it is thought that the BBB restricts access into the “immune-privileged” central nervous system (CNS) to limit potentially deleterious inflammation. This represents a possible point of therapeutic intervention in treating leukocyte driven diseases of the CNS like multiple sclerosis. Tight junctions are thought to be a more formidable barrier to diapedesis than adherens junctions and, correspondingly, transendothelial migration into this privileged tissue could occur preferentially through the endothelial cell body (transcellular migration). This would allow the leukocyte to bypass the junction and avoid its disruption. Indeed, there are reports suggesting that transcellular migration predominates in this context [33–35]. It remains unclear if this is so; however, it is also unclear whether mechanisms exist, if any, to remodel the tight junctions and permit paracellular TEM.
Migration beyond the endothelium
After passing across the endothelial cell layer, leukocytes must then navigate the basement membrane and extracellular matrix that is deposited by both endothelial cells and pericytes. This complex mesh-like network of fibrils that provides support for the endothelial cells presents a formidable barrier because it is interconnected by covalent bonds and cannot be remodeled rapidly and reversibly like the VE-cadherin at the endothelial cell borders. Indeed, in vivo observations of leukocyte migration often report that the migration through the basement membrane takes a relatively long time, requiring tens of minutes instead of the few minutes required for TEM [36]. To get through this barrier, leukocytes have been observed to migrate through areas with a low density of ECM components and pericytes [37]. Relatively little is known about the processes and molecular interactions involved, but it is an area of intense research. The process has been the topic of some excellent reviews to which the reader is referred for further information [38, 39].
The molecules involved on the endothelial cell
ICAM-1 and VCAM-1
While selectins play an important role in the initial steps of the adhesion cascade (reviewed in detail elsewhere [3, 5, 40]), most of the downstream activation and migration steps are mediated by several members of the immunoglobulin (Ig) superfamily of adhesion molecules [41, 42]. Intercellular adhesion molecule-1 (ICAM-1, CD54) and vascular cell adhesion molecule-1 (VCAM-1, CD106) both act downstream of the selectin-mediated loose adhesion step and function in facilitating strong adhesion [43]. ICAM-1 and VCAM-1, which are expressed on the endothelial cell, bind integrins on the leukocyte. ICAM-1 binds to both Lymphocyte Function-Associated Antigen 1 (LFA-1, αLβ2, a dimer of CD11a and CD18) and Macrophage-1 Antigen (Mac-1, αMβ2, a dimer of CD11b and CD18) whereas VCAM-1 binds to Very Late Antigen-4 (VLA-1, α4β1, a dimer of CD49d and CD26). ICAM-1 and VCAM-1 binding to these integrins triggers a series of critical signaling events in both the endothelial cell and leukocyte. Both ICAM-1 and VCAM-1 are expressed diffusely on the endothelial surface but become enriched under the leukocyte as it moves across the endothelial cell [44, 45]. The enrichment of ICAM-1 is dependent on the actin cytoskeleton and remodeling events that include the Src-mediated phosphorylation of cortactin, an actin binding protein [32, 46, 47]. VCAM-1 clustering activates the small GTPase Rac-1 [48, 49] and triggers the release of intracellular calcium [50, 51] and recruitment of ezrin and moesin [52]. These proteins, together with actin, ICAM-1, and VCAM-1, have been reported to form dynamic “docking” structures or transmigratory cups [45, 53]. These structures appear as finger-like protrusions that rise up from the endothelial cell and surround the leukocyte and are dependent on proper ICAM-1 and VCAM-1 activation and function. Treatments with cytochalasin D and colchicine abolished their formation indicating a dependence on an intact actin and microtubule cytoskeleton [53, 54]. Interestingly though, these docking structures are not observed around all leukocytes or under all conditions [55, 56]. They have only been identified in vitro and appear to form under conditions where leukocyte migration is delayed or protracted and disappear when TEM resumes, possibly indicating the exaggeration of a normally minimal process [52, 54, 57]. Likewise, transmigratory cup formation could occur preferentially during transcellular TEM possibly indicating a specialized role in what has been assumed to be a more difficult and less frequent process [19, 54].
ICAM-2
ICAM-2 was first identified in a biochemical screen for novel LFA-1 ligands [58]. It is a 55-kDa Ig superfamily proteoglycan that has 35 % similarity to ICAM-1. ICAM-2 is expressed on endothelial cells, and, to a lesser extent, leukocytes and platelets. However, unlike ICAM-1 which is diffuse across the endothelial apical surface, ICAM-2 is localized to endothelial cell borders and is expressed at much higher levels in high-endothelial venules [59]. While ICAM-1 expression is induced upon treatment with the inflammatory cytokines TNFα or IL-1β, ICAM-2 express is down regulated, possibly indicating a role in TEM into non-inflamed tissues [60, 61]. Studies of the role of ICAM-2 in inflammation have produced mixed results regarding its importance. In an allergic asthma model of inflammation, ICAM-2 knockout mice show a defect in the recruitment of eosinophils, but not lymphocytes or monocytes [62]. The requirement for ICAM-2 could be highly dependent on the inflammatory stimulus as it was shown to be required for neutrophil recruitment in an IL-1β peritonitis model but not in TNFα- or thioglycollate-induced responses [63]. This could also be due to functional redundancy with ICAM-1 as function blocking antibodies against ICAM-2 block leukocyte recruitment in an ocular model of inflammation but only upon knockout of ICAM-1 [64]. Although this would suggest overlapping functions between ICAM-1 and ICAM-2, several reports have found that ICAM-2 plays a more critical role in cell polarization and leukocyte migration on the endothelium while ICAM-1 is primarily involved in adhesion [63, 65]. Together, these findings suggest that the role of ICAM-2 in inflammation may be highly dependent on the specific inflammatory context in which it is examined.
Junctional adhesion molecules
Although they have been primarily studied in the related process of leukocyte migration across the epithelial barrier, several members of the Junctional Adhesion Molecule (JAM) family have been reported to be involved in TEM [66, 67]. The JAM family consists of three closely related proteins (JAM-A, -B, and -C) sharing ∼35 % sequence similarity and two distantly related members (JAM-4 and JAM-L) which have only ∼16 % similarity to the first three. JAM-4 and JAM-L have not been investigated thoroughly in TEM and consequently will not be discussed here. All JAM members are membrane proteoglycans and contain two extracellular Ig-domains which facilitate both homophilic and heterophilic interactions [66, 68]. While JAM-A, -B, and -C are all expressed on endothelial cells along the cell borders, JAMs -A and -C are also expressed on most leukocyte subtypes, but only JAM-A is expressed on epithelial cells [66]. JAM-A appears to be a ligand for LFA-1 [69]. Antibody-mediated disruption of this interaction abrogates the ability of endothelial cells to support the adhesion and TEM of neutrophils in vitro [69] and monocytes in vitro and in vivo [70, 71], although in other in vitro assays, its role in TEM is less clear [72]. JAM-B has been reported to bind VLA-4 [73] whereas JAM-C binds Mac-1 [74]. Although the function of these interactions in TEM has not been fully investigated, antibodies against JAM-C and soluble JAM-C extracellular domain both decreased leukocyte emigration in vivo [75–77]. Recent reports also suggest that JAM-B may interact with JAM-C to support its function in TEM [78, 79].
PECAM
Platelet/endothelial cell adhesion molecule-1 (PECAM, also known as CD31) is a ∼130 kDa Type 1 membrane glycoprotein that belongs to the Ig superfamily. PECAM is expressed diffusely on leukocytes and platelets but in endothelial cells it is enriched at the cell border [80]. It has six extracellular Ig repeats and contains several known and putative phosphorylation sites including two intracellular immunoreceptor tyrosine-based inhibitory motifs (ITIM) [81]. During TEM, PECAM on the leukocyte and endothelial cell bind homophilically [82, 83]. Disruption of this interaction using genetic ablation or function blocking reagents (PECAM-Fc chimeras or antibodies that bind domain 1 of PECAM) arrests leukocytes on the apical surface of the endothelial cell above the junction. When visualized live, these arrested leukocytes are not stationary but instead migrate up and down along the junction as if poised to undergo TEM but lacking a critical signal [84].
Poliovirus receptor
Poliovirus Receptor (PVR, also known as CD155) was originally identified based on its role as the key receptor for poliovirus entry into cells [85]. More recently, it has also been found to be involved in cell adhesion and motility [86–89], T-cell development [90, 91], platelet activation [91, 92], NK and CD8 T-cell cytotoxicity [93, 94], and TEM [95–97]. PVR is a transmembrane Ig superfamily proteoglycan expressed on neurons, epithelium, endothelium, and monocytes/macrophages. PVR, like PECAM, contains an ITIM motif, although not much is known about the role of this domain in the context of PVR function in TEM [87]. PVR localizes to sites of cell–cell and cell–matrix adhesion and binds to DNAM-1 to facilitate a diverse array of functions [85, 97–102]. DNAM-1 is a transmembrane immunoglobulin superfamily member expressed on NK cells, T-cells, some B-cells, platelets, monocytes, and possibly activated human umbilical vein endothelial cells (HUVEC) [90, 91, 93, 103, 104]. Reymond et al. demonstrated a role for the molecules PVR and DNAM-1 in TEM, showing that monoclonal antibodies against either molecule could block the transmigration of isolated monocytes across a monolayer of cultured HUVEC [97]. More recently, Manes and Pober used antibodies against PVR and DNAM-1 to block TEM of effector memory T-cells [96]. However, characterization of the mechanism of action and the relationship of these molecules to other components of the TEM machinery was only recently uncovered (discussed below) [95].
CD99 and CD99L2
CD99 is a small 32-kDa Type 1 membrane protein that is highly O-glycosylated [105]. It does not belong to a characterized superfamily and only exhibits sequence similarity to one other protein, CD99 antigen-like protein 2 (CD99L2). CD99L2 is a 45-kDa Type 1 membrane protein that has 32 % amino acid similarity to CD99 and is similarly highly O-glycosylated [106, 107]. Both have short (<40 amino acids) but divergent cytoplasmic domains. Like PECAM, CD99 and CD99L2 are expressed at endothelial cell junctions and diffusely on leukocytes. CD99 facilitates TEM through homophilic interactions between the two cell types [108]. Similarly, CD99L2 has been reported to function through homophilic interactions but it may have another unidentified ligand [109–111]. Disruption of CD99 and CD99L2 interactions using function blocking antibodies or genetic knock out (in the case of CD99L2) impairs leukocyte extravasation in vitro and in vivo. Interestingly, unlike PECAM blockade, which arrests leukocytes on the apical surface, blocking CD99 function in vitro traps the migrating leukocytes midway through the junctions [108, 112]. While the requirement for CD99 and CD99L2 in TEM is established, the mechanism by which they control TEM is unknown. In mice, blocking either CD99 or CD99L2 by polyclonal antibody arrested leukocytes in vivo a similar step in extravasation, suggesting that the two proteins functional to facilitate the same step [113].
Although there are several regions that are highly conserved between the two molecules and across species, there are no known relevant interactions with other proteins that have been reported. Interestingly though, in a study using the mouse homologues of CD99 and CD99L2, Nam and coworkers showed that the two molecules interact with each other heterophilically through their cytoplasmic tails [111]. Through this interaction CD99 appears to facilitate the trafficking of CD99L2 to the plasma membrane.
Key events in TEM
Sequential functions of molecules in TEM
One interesting finding that is beginning to be appreciated in the field is that several of these molecules have been observed to function in a sequential manner during TEM [18, 114, 115]. Current understanding of the process is that ICAM-1 and VCAM-1 function upstream of PECAM and CD99. This finding is well supported in the literature both in vivo and in vitro [55]. It also fits with the subcellular localization of these proteins with ICAM-1 and VCAM-1 both localized to the apical surface of endothelial cells where they can function in the activation and adherence of captured leukocytes whereas PECAM and CD99 are localized to the cell border where they facilitate the subsequent migration though the junction.
Furthermore, antibody blockade studies have highlighted an additional level of sequential function for PECAM, PVR, and CD99, operating in that order [95, 108]. These assays took advantage of the observation that the antibody blockade of one molecule can be relieved by extensive washing of unbound antibody and allowing additional time for TEM to recover. For example, incubating transmigrating leukocytes with anti-PECAM antibodies halts TEM at the PECAM-requiring step. After washing out the antibody, the arrested leukocytes can then resume and complete TEM. If instead antibodies against CD99 are added during the recovery phase, the leukocytes remain arrested, unable to complete transmigration [108]. However, reversing the order of the antibody treatments (anti-CD99 initially, anti-PECAM during recovery) yielded no net block. For this combination, the leukocytes were presumably able to migrate past the step requiring PECAM during the first incubation and thus the anti-PECAM antibodies had no effect during the recovery phase, allowing the leukocytes to complete TEM. Taken together, these results indicate that PECAM works upstream of CD99. Using the same methodology, it was recently found that PVR functionally interacts with DNAM-1 at a step in between those regulated by PECAM and CD99 [95]. Thus, based on these in vitro studies, the net order of events is ICAM-1 and/or VCAM-1 interacting with their leukocyte integrin partners on the apical surface followed by PECAM, PVR/DNAM-1, and CD99 for diapedesis per se.
The sequential control of TEM is apparently much more complex in vivo. Various investigators report differences in the degree to which blocking specific molecules results in reduction of TEM and the relative order in which these molecules function. Some of these seemingly conflicting results can be reconciled by differences in the inflammatory stimulus, tissue and mouse strains used in the various studies. In most mouse strains examined, PECAM deficiency or blockade arrests leukocytes on the apical side of the endothelium, consistent with in vitro findings using human cells [116]. In the C57BL/6 strain however, depending on the stimulus, PECAM knockout has no effect on leukocyte TEM and instead leads to leukocyte arrest at the basement membrane, suggesting an additional role for PECAM (in particular extracellular domain 6) in the inflammatory process [117]. Antibody blockade of ICAM-1 decreased both adhesion and transmigration whereas ICAM-2 blockade only inhibited transmigration, suggesting that ICAM-1 functions upstream of ICAM-2 [63]. To complicate the matter further, the requirement ICAM-2 was only observed in an IL-1β but not in TNFα or thioglycollate model of peritonitis [63]. In a follow-up study, also done in the C57BL/6 background, PMN migration in response to TNFα was unaffected by knockout of ICAM-2, JAM-A, or PECAM mice [118]. However, neutrophils that lacked TNFR show significantly reduced infiltration in these mouse strains suggesting roles of these molecules are stimulus-dependent. Furthermore, examination of the site of arrest in the ICAM-2−/−, JAM-A−/−, and PECAM−/− mice in the C57Bl/6 strain showed that neutrophils were arrested on the endothelial surface, partway through the junction, and at the basement membrane respectively, suggesting that these molecules function in that order [118]. Also in the C57BL/6, Vestweber and coworkers showed that disruption of CD99, CD99L2, and PECAM function in IL-1β models of inflammation all caused migrating leukocytes to be arrested in the same place, at the level of the basement membrane [113]. Interestingly though, in TNFα-induced inflammation, only CD99 and CD99L2 disruption caused a block in TEM, suggesting under this particular set of conditions, these two molecules function in a different pathway than that in which PECAM functions.
The LBRC
When examined by standard wide-field immunofluorescence, all of the endothelial proteins known to be critical to TEM are predominantly localized to the endothelial cell border. At this resolution, the expression pattern makes it impossible to determine if they actually co-localize on a molecular scale under resting conditions. During TEM, several of these molecules have been observed to move in divergent ways. In short, whenever they have been examined, the molecules that have cognate interacting partners on the migrating leukocyte become enriched around the leukocyte whereas molecules that are responsible for maintaining junctional integrity appear to move out of the way [25, 119, 120]. Thus, for a leukocyte in the process of paracellular TEM, the endothelial pools of adhesion molecules like PECAM and JAM-A are enriched around the leukocyte and a ring is observed in addition to the regular junctional localization. Adherens junction proteins like VE-cadherin, on the other hand, are absent from the site of TEM and thus a gap appears in the usual staining pattern [17, 121].
One possible explanation for the observation that adherens junction proteins are excluded from the TEM site whereas adhesion molecules like PECAM are enriched is that there could be two separate pools or domains containing these molecules at the cell border. To investigate this closer, the subcellular localization of these proteins was examined using electron microscopy (EM) and horseradish peroxidase (HRP)-conjugated monoclonal antibodies. VE-cadherin antibodies showed the expected staining along the endothelial border (Fig. 1). Similarly, PECAM antibodies also showed localization at cell–cell contacts, but unlike VE-cadherin, PECAM was also observed in a subjunctional grape-like structure of interconnected 50 nm vesicles and tubules [119] (Fig. 1). PECAM from this compartment was observed to exchange and recycle with the surface membrane (discussed below) and, consequently, termed the Lateral Border Recycling Compartment or LBRC [119]. The limiting membrane of the LBRC is contiguous with the plasma membrane, suggesting that this compartment is a complex invagination of the junctional membrane. Positively stained clusters were observable several vesicle diameters away from the cell border suggesting that the LBRC extends several hundred nanometers into the cell. Interestingly, the compartment is labeled well at 37 °C, but incubating endothelial cells with the same antibodies at 4 °C only labeled the junction [95, 119]. Electron micrographs clearly show that the compartment still exists under this condition, indicating that PECAM in the LBRC is protected from large molecules at 4 °C. This is not because the vesicles have pinched off, as protons and small molecules can still enter ([119] and unpublished results). Under resting conditions roughly 30 % of the total PECAM is in the LBRC [119]. This compartment also contains JAM-A [55] and CD99 [55] (Fig. 1), PVR [95], and nepmucin [122], which regulates lymphocyte TEM in high-endothelial venules. The LBRC is also observed in vivo as EM examination blood vessels from mice injected intravenously with anti-mouse PECAM-HRP shows an identical subjunctional staining pattern (unpublished results).

PECAM, CD99 and JAM-A, but not VE-cadherin are in the lateral border recycling compartment. Endothelial cells were incubated with HRP-conjugated mAb specific for PECAM, CD99, JAM-A, or VE-cadherin for 1 h. at 37 °C, then fixed and reacted with diaminoabenzidine-H2O2 as described [55, 119, 123]. As a control for nonspecific labeling, free HRP was added at the concentration present on the antibodies. En face sections were cut for EM analysis. In addition to being present along the cell border (dark electron-dense staining) PECAM, CD99, and JAM-A are all present in interconnected vesicular structures of the LBRC. VE-cadherin is present at the cell border (arrowhead), but not in the interconnected vesicular structures adjacent to it (arrows). Arrowheads indicate cell borders; arrows indicate the LBRC. Scale bar 200 nm. Reproduced from [55] with permission
To determine the kinetics of entry and turnover in the LBRC, endothelial cells were treated with unconjugated Fab fragments of non-blocking antibodies at 37 °C to bind to total (surface + internal compartment) PECAM. The endothelial cells were then incubated with unconjugated secondary antibodies at 4 °C to saturably bind all the labeled PECAM, but only at the junctional surface. After washing and warming the monolayer in the presence of fluorescent secondary, the once protected PECAM-Fab complexes can then be labeled as the LBRC recycles to the surface. When viewed using immunofluorescence microscopy, the time-dependent increase in fluorescence at the junction (representing the increased accessibility of the once hidden epitope) is readily observed with a half-time of ∼10 min [119]. These results suggested that the compartment undergoes continuous exchange or recycling of its components.
Because the anti-PECAM-Fab fragment does not block TEM, the labeling procedure described above can be used to follow the LBRC during TEM. When leukocytes are allowed to migrate for short time points (∼8–10 min, to catch them in the act of TEM) on endothelial cells labeled in this fashion, a significant increase in fluorescence (representing PECAM from the once protected LBRC) is observed as a ring around the migrating cell [119] (Fig. 2). The mean intensity of the fluorescence in the ring is roughly three- to fivefold higher than the fluorescence observed at the junction indicating that the LBRC is specifically directed to and enriched at the site of TEM. This specific enrichment around the transmigrating leukocyte is referred to as “targeted recycling” of the LBRC.

Targeted trafficking of membrane from the LBRC to surround leukocytes undergoing transcellular migration. PECAM in the LBRC was labeled with Fab fragments of a non-blocking mAb as a surrogate marker for LBRC trafficking to the endothelial surface, as described [55, 119, 123]. Staining of recycled PECAM as a surrogate for recycling LBRC (left panel, red in overlay) surrounding monocytes (upper panel) and a PMN (lower panel) demonstrates movement of membrane from the LBRC to the endothelial surface. Recycled PECAM is seen around the leukocytes migrating transcellularly (thin arrows) far from the endothelial border (arrowheads). Recycling LBRC enrichment is as great around the monocyte migrating transcellularly as it is around the monocyte migrating paracellularly (thick arrow). Actin staining (middle panel, green in merge) shows relative positions of the cells. Orthogonal (X–Z) projection demonstrates leukocytes in the act of transcellular migration. Arrows indicate recycled PECAM around leukocytes. Bar 10 μm. Reproduced from [55] with permission
During TEM, the LBRC surrounds the migrating leukocyte and there is growing evidence that this is a critical step in diapedesis. Disrupting PECAM–PECAM homophilic interactions ablates both TEM and targeted recycling [119]. The enrichment of the LBRC at the leukocyte is similarly blocked when the endothelial cells are treated with reagents demecholcine or taxol, which depolymerize microtubules or cause microtubule bundling, respectively [123]. Neither treatment affected the size or the constitutive recycling of the compartment suggesting instead that microtubules are essential for LBRC movement during TEM. This is supported by the finding that LBRC targeted recycling is also impaired upon microinjection of function blocking antibodies against the motor domain of kinesin, a molecular motor that facilitates intracellular traffic along microtubules [123]. In fact, it appears that the directed movement of the LBRC is the critical factor to support TEM as the migration of lymphocytes, which exhibit PECAM-independent migration [124, 125], is abolished using these treatments that disrupt LBRC targeted recycling [123]. Together, these findings indicate that the LBRC is directed to the site of TEM using microtubules and kinesin to provide the migrating leukocyte with functional pools of the molecules required for it to undergo TEM.
The presence within the LBRC of so many molecules that are involved in the endothelial cell’s role in TEM suggests that it could act as a reservoir of these molecules in TEM. More important, the available data suggest that the molecules in the LBRC, not those on the surface of the junction are the ones that are required for TEM. When endothelial cells were treated with blocking antibodies against PECAM at 4 °C, which leaves the protected LBRC pool of PECAM untouched, TEM remained unaffected, while the same antibodies given at 37 °C blocked TEM [82]. More recently, and using the same methodology, it has become clear that the portions of CD99 and PVR that are residing in the LBRC are the functionally relevant pools that are required for TEM [95].
It is important to note that the LBRC is distinct from other reported endothelial vesicular organelles such as the vesicular vacuolar organelle (VVO) and caveolae. VVOs are much larger (>150 nm) in size, more heterogeneous in shape, and typically communicate between the apical and basal surfaces [126]. VVOs open and close in response to VEGF whereas the LBRC is completely and continuously accessible to the exterior of the cell [127]. VVOs have not been observed in vitro. Likewise, caveolae are rare in endothelial cells in vitro and are typically observed as single vesicles or single invaginations on the apical and basal membranes, seldom present at the junction. By immunofluorescence, caveolin-1 (a marker of both caveolae and VVOs [128, 129]) does not co-localize with PECAM at the junction or during TEM [55, 119]. In addition, biochemical analysis of PECAM and caveolin-1 shows that the two localize to different membrane microdomains [119]. This was examined using sucrose gradients to purify cholesterol-rich membrane microdomains from endothelial cells solubilized with cold non-ionic detergent [119]. In this analysis, caveolin-1 was predominately recovered in the buoyant fractions that correspond to lipid rafts whereas PECAM was recovered from the dense fractions which contained the solubilized proteins that are excluded from membrane microdomains (this is in distinction to leukocytes, where activation of PECAM led to its phosphorylation and partition into cholesterol-rich microdomains [130] and in platelets where a small fraction of palmitoylated PECAM can be recovered in such domains [131].)
Although several molecules are known to be partially localized to the LBRC, almost nothing is known about the signals the meditate entry or inclusion into the compartment. Analysis of the cytoplasmic tails of PECAM, CD99, JAM-A, and PVR does not reveal any of the known sorting signals, like the well-defined KDEL or DXXLL sorting signals used in the canonical secretory pathway [132–134]. Likewise, none of the proteins show any obviously conserved regions or amino acids that could serve as novel sorting motif. To date, the only known instance of altered inclusion into the LBRC was observed using PECAM phosphorylation mutants in the cell line, ECV304 [135]. ECV304 cells lack PECAM expression and do not support TEM. However, this TEM defect can be rescued when exogenous PECAM is expressed, making it a convenient system to test PECAM function. ECV304 cells expressing PECAM exhibited targeted recycling of the LBRC. Examination of the critical tyrosines in the ITIM motif of PECAM showed that the mutation of Y663 to phenylalanine (Y663F), but not Y686F, abolished TEM and targeted recycling of the LBRC [135]. Constitutive recycling of PECAM Y663F was also attenuated, indicating that this residue is either critical for PECAM sorting into the LBRC or that it significantly disrupts LBRC structure of function. In support of this, a more detailed analysis comparing the total pool of PECAM and that PECAM that is protected at 4 °C shows that virtually all of the phosphorylated PECAM resides in the LBRC [136]. Blocking this phosphorylation with the src-family kinase inhibitory PP2 or the broad spectrum tyrosine phosphorylation inhibitor Genistein prevents both targeting recycling of the LBRC and TEM [136]. PVR can also be phosphorylated on a tyrosine in its ITIM in a src-dependent manner [87, 137]. Under resting conditions, very little PVR is phosphorylated, however when PVR is stimulated with antibodies or with DNAM-1-Fc chimeric protein (to simulate the interaction that is relevant to TEM) PVR becomes phosphorylated [95]. Together, these data suggest that phosphorylation of these molecules may play a critical role in their localization and function in the LBRC during transmigration.
Transcellular TEM and the LBRC
Transcellular TEM (migration through the endothelial cell body away from the junction) is a distinct process from paracellular TEM at the junction [138, 139]. In most reports, paracellular TEM is the predominant route with transcellular migration reported occurring in less than 10 % of events, both in vitro and in vivo. Transcellular TEM is thought to occur more often at the BBB, where intercellular junctions are more complex [35, 140], and also in situations with highly activated endothelial cells and leukocytes [53, 54]. During transcellular TEM the enrichment of PECAM and the other LBRC components around the leukocyte is still observed (Fig. 2) [55, 141]. Obviously there are key biological differences between the two methods; junctional remodeling would only be necessary for paracellular TEM whereas only transcellular TEM would require the de novo creation of a conduit between the apical and basal surfaces. In spite of these differences, there are several similarities between the two. Disrupting PECAM or CD99 interactions also inhibits transcellular migration [55]. Targeted recycling of the LBRC is required for both routes and in each case it can be inhibited by disrupting microtubules or kinesin function [55]. Interestingly, although cells treated with microtubule depolymerizing drugs do not show enriched PECAM fluorescence under the leukocyte and do not support transcellular migration, they are still able to enrich ICAM-1 under the leukocyte. This shows that ICAM-1 functions at a step that occurs early in leukocyte recruitment and is therefore not dependent on LBRC or PECAM function [55].
Thus, it appears that the LBRC has a nearly identical role in transcellular migration as in paracellular TEM. It is directed to meet and deliver critical molecules (largely absent from the apical surface) to the migrating leukocyte. Furthermore, because of its interconnected reticular structure, it contains a significant amount of membrane to help facilitate the formation of a transcellular pore. In theory, this model would require, at most, one membrane fusion event (the LBRC fusing with the plasma membrane in the cell body) at the apical surface and one at the basal surface. Although the mechanism that mediates this event is not currently known, it could be accomplished using the same SNARE proteins that facilitate many other membrane fusion events [142, 143]. Several of these events are triggered by local increases in calcium signaling and the clustering of ICAM-1 and VCAM-1 has been shown to signal to other molecules using calcium. Indeed, inhibition of SNARE activity or buffering endothelial calcium both significantly inhibit transcellular migration [141].
Signaling events related to TEM
Phosphorylation and dephosphorylation
Although several lines of evidence have shown a connection between vascular permeability and the global phosphorylation of adherens junction proteins, it has only recently become appreciated that similar mechanisms are at work in the context of TEM. A few of the pathways could also function in the recruitment and function of the LBRC and will be discussed briefly here. For a more comprehensive analysis of mechanisms, the reader is referred to [22, 144] and the other chapters in this volume. Crosslinking E-selectin or ICAM-1 or adhesion of leukocytes induces outside-in signaling in endothelial cells and, among other things, activates src kinase [145]. Crosslinking of ICAM-1 also activates the kinase pyk2 [28]. Cortactin was identified as a substrate for src and its phosphorylation is critical to the clustering of ICAM-1 and its association with the actin cytoskeleton [46, 47, 146]. Disruption of this process significantly impaired TEM but did not affect adhesion. The role of src does not stop there, however; ICAM clustering has since been shown to lead to the phosphorylation of VE-cadherin via the activities of both src and pyk2 [28]. Inhibition of this phosphorylation, either using specific inhibitors or tyrosine-to-phenylalanine VE-cadherin constructs that cannot be phosphorylated, similarly reduced TEM [28]. The phosphorylation of VE-cadherin disrupts its interaction with p120 and β-catenin, which anchor it to the actin cytoskeleton. The interpretation of these results is that the dissociation of VE-cadherin from the cytoskeleton at the site of TEM allows the junction to part for the migrating leukocyte to pass through. This is supported by the finding that over-expression of p120, which stabilizes VE-cadherin at the junction, significantly reduced VE-cadherin phosphorylation and impaired PMN TEM [25, 120]. As mentioned above, src kinase phosphorylation of PECAM is required for entry into and targeted recycling of the LBRC [135, 136] whereas PVR is phosphorylated by src upon ligand stimulation with DNAM-1 or crosslinking [95]. While it is not clear yet if these later events are directly linked to upstream events like E-selectin or ICAM-1 stimulation, these collectively point to a critical role for src in the process of TEM.
In what appears to be analogous recruitment scheme, the phosphatase SHP2 has been reported to be similarly recruited to several of the molecules involved in TEM. Poluskota and coworkers identified a loosely conserved ITIM motif in the cytoplasmic tail of ICAM-1 that is phosphorylated and mediates the recruitment of SHP2 upon ICAM-1 stimulation [147]. Although this study used fibrinogen to activate ICAM-1 instead of the more classical antibody-mediated crosslinking, they are supported by the finding that the SHP2 plays a role in ICAM-1 signaling. Minshall and coworkers have recently reported that SHP2 dephosphorylates src tyrosine 530. In doing so, it removes an inhibitory phosphate that blocks the activity of src. The expression of a dominant-negative form of SHP2 prevented the src activation upon ICAM-1 crosslinking indicating that this dephosphorylation event is required for ICAM-1 mediated signaling [148, 149]. SHP2 also plays a role in VE-cadherin and adherens junction remodeling. SHP2 associates with β-catenin in VE-cadherin/p120/β-catenin/plakoglobin complexes. Ablation of SHP2 expression in endothelial cells causes increased phosphorylation of all of the components in the complex and, consequently, increased permeability [150]. Although these experiments were performed using thrombin treatment, which affects global junctional integrity, it is tempting to speculate that similar mechanisms might be at work in on a local scale during TEM. In further support of this are the findings that SHP2 in endothelial cells also associates with the phosphorylated ITIM motifs of both PECAM [135, 151, 152] and PVR [95, 137], although the downstream functions of this association in LBRC function have yet to be determined.
Other signals regulating TEM
The VCAM-1 and ICAM-1 mediated changes in VE-cadherin phosphorylation have been shown to occur through the activation of several other signaling pathways. VCAM-1 clustering leads to the activation of MAP kinase (MAPK) and the GTPases Rac1 and RhoA, and induces the production of actin stress fibers and reactive oxygen species (ROS) [49, 153]. Similarly, ICAM-1 crosslinking activates RhoA and the clustering of either VCAM-1 or ICAM-1 can cause transient increases in intracellular calcium [51]. Signaling through these pathways has been shown to cause disruption of adherens junction complexes and retraction of intercellular contacts, which leads to increased endothelial permeability. Several of these pathways may be acting on a more local scale to “loosen” the junction and facilitate TEM. Activation of Rho induces the phosphorylation of myosin light chain through the activation of myosin light chain kinase (MLCK) [154, 155]. Inhibition of either Rho or MLCK has no effect on leukocyte adhesion but substantially blocks TEM [49, 155].
Addition of activated leukocytes to activated endothelium induces a transient increase in cytosolic calcium that is observed after adhesion but preceding TEM, and may occur through the activation of selectins and/or ICAM/VCAM [51, 156–158]. Indeed, buffering intracellular calcium does not affect adhesion but blocks neutrophil [159] and monocyte TEM [156]. Likewise, VCAM-1 stimulation also induces Rac-mediated ROS production through the activity of nicotinamide adenine dinucleotide phosphate oxidase (NOX) [30, 49]. The activation of Rac and NOX lead to activation of pyk2 which subsequently results in the dissociation of VE-PTP from VE-cadherin, which is a critical step in opening the junctions to allow for TEM [30]. When any of the steps in this pathway are blocked, VE-PTP remains associated with VE-cadherin and leukocyte TEM is attenuated [29, 30, 49]. Similarly, ICAM-1 stimulation leads to phosphorylation of endothelial nitric oxide synthase (eNOS) and the production of nitric oxide [153]. Signaling through the eNOS pathway has been shown to be required for leukocyte TEM both in vitro [153] and in vivo [149]. Although this growing body of evidence suggests that these pathways are important to TEM, much remains to be determined about how they fit into the overall process.
How might the LBRC be doing its function?
Given that several of the molecules and signaling pathways that are activated or recruited during upstream events like ICAM-1/VCAM-1 clustering also have roles in TEM events like LBRC recruitment and junction remodeling, it is possible that the prelude to TEM (e.g., leukocyte adhesion and locomotion on the endothelial surface) primes these signaling pathways for rapid function to facilitate TEM. The recruitment of SHP2 activates Src (by removing the inhibitory phosphorylation of src Y530) which can then phosphorylate cortactin and reinforce ICAM-1 clustering. Src may also then phosphorylate ITIM motifs of PECAM and PVR, brought to the site of TEM by the targeted recycling of the LBRC. If the migrating leukocyte is near a junction, VE-cadherin and its associated adherens junction protein would be phosphorylated, leading to a local disassembly of the junctional barrier. The leukocyte, now presented with both the plethora of unligated adhesion molecules delivered from the LBRC and a loosening of the junction, could extend pseudopods through the opening and migrate across the endothelium. After the leukocyte has passed, reforming the adherens junction could be aided through the activity of SHP2 brought to the TEM site by PECAM and PVR. After dephosphorylation of VE-cadherin and its associate proteins, stable adherens junctions could reestablish their connection across the junction and with the actin cytoskeleton, and the whole mechanism would be reset for another round of TEM.
During transcellular migration, the same processes would function except that the LBRC itself serves as the conduit allowing passage across the endothelium in place of a gap at the junction. In both cases, the LBRC, serving as a repository of several critical molecules, functions in TEM by delivering these molecules en mass to the site of TEM. It is tempting to speculate that the LBRC serves as a platform for the recruitment of key cytosolic signaling molecules as well. In doing so, the endothelial cell would then only have to recruit one compartment instead of recruiting each molecule individually, which would be mechanistically more complicated.
The LBRC could also function to help solve another potential issue concerning the physics of junctional remodeling and the shape changes that the endothelial cell must undergo during TEM. Although endothelial cell contacts are serpentine when viewed on a large scale (i.e., over the whole periphery), at the smaller micron scale that is particularly relevant to TEM they are relatively straight, i.e., over the course of a few microns they do not meander greatly. Thus, in order for the leukocyte to pass, not only must the endothelial cell move VE-cadherin and other junctional molecules out of the way, but it must also supplement the surface area of the membrane to accommodate the requisite pore. To appreciate the magnitude of this problem, the approximate amount of surface area required can be estimated from a few simple assumptions. The numbers in this example were chosen for simplicity, but the example would work for any reasonable pore size. Assuming that the pore that forms during TEM is approximately 3 μm in diameter, the thickness of the endothelial cell is roughly 1 μm at the border, and the junction is perpendicular to the plane of the endothelium, the surface area of the each membrane at the future site of the pore is 3 μm2 (length (l) of the junction times the height (h)), making the total surface area of the two borders together 6 μm2. The total surface area around the pore however would be ∼9 μm2 (modeled as a cylinder with a diameter (d) of 3 μm and a height (h) of 1 μm). This rough approximation suggests that the membrane surface area at the site of TEM must locally increase roughly 50 %. While this need could be met by a simultaneous straightening of a distant portion of undulating lateral membrane, this would likely require cytoskeletal rearrangements that would likely be difficult to coordinate with the rapid process of TEM. However, the LBRC, being a contiguous invagination of the lateral membrane distributed around the entire cell periphery, is ideally localized to provide this needed surface at the moment of TEM. Approximately one third of the total PECAM and CD99 in endothelial cells is in the LBRC, so recruitment of LBRC membrane would add this additional 50 % surface area. Therefore, this compartment is also the appropriate size to contribute this amount of membrane to the cell border.
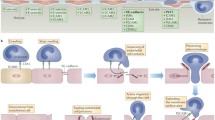
This theory has additional appeal when considered alongside the proposed mechanism for removing VE-cadherin from the TEM site. Various hypotheses have been entertained regarding this topic (Fig. 3). Some early reports suggested that VE-cadherin is degraded by surface proteases on leukocytes during TEM (Fig. 3b) [160, 161], however this has not been supported in more recent findings. One current opinion is that VE-cadherin is either internalized into endocytic vesicles (Fig. 3c) in a phosphorylation dependent manner, akin to the internalization observed upon global treatment of endothelial cells with reagents that increase vascular permeability [162, 163]. However, biochemical studies have not detected change in the amount of VE-cadherin that is protected in endosomes during transmigration [25]. Similarly, using live cell microscopy, Luscinskas and coworkers could not detect VE-cadherin-GFP accumulation in vesicles during the formation of a TEM pore [25]. These findings suggest instead that VE-cadherin, after dissociating from the actin cytoskeleton, is moved laterally to the membrane adjacent to the nascent pore (Fig. 3d). This lateral movement could be facilitated by the addition of membrane from the LBRC, i.e., the LBRC is targeted to the TEM site and by incorporating its lipid and protein content, VE-cadherin is shunted off to the side out of the way (Fig. 3e). This model is particularly attractive when considering that the other mechanisms do nothing to increase the local surface area at the TEM site and internalization of VE-cadherin would actually decrease it locally. After TEM is completed, the LBRC could be retracted back into its interconnected structure allowing VE-cadherin to repopulate the gap, connect with the actin cytoskeleton and return the junction to the basal state.

Schematic representation of the VE-cadherin gap formation at the junction. a Under resting conditions, VE-cadherin (red bars) interacts homophilically across the junction and connects to the cytoskeleton and the lateral membranes of two adjacent cells (shown in blue) in close proximity. The schematic of the junction is shown as a cross section when viewed from above. Consider that this junction would extend into the third dimension and would have an initial surface area defined as SAi for simplicity. During TEM, VE-cadherin is removed from the junction at the site of the transmigrating leukocyte. This could occur by b degradation of the extracellular domain, c internalization into vesicles, d lateral movement to adjacent areas, and/or e recruitment of the LBRC and displacement of the existing membrane. Only the recruitment of the LBRC would locally increase the membrane surface area at the TEM site and allow for efficient formation of a transmigration pore (f)
Conclusion
The migration of leukocytes out of the blood stream is a complex process that requires the careful coordination of diverse signaling events. Several of these pathways are initiated by the clustering of the selectins, ICAM-1, and VCAM-1 in the early steps of leukocyte capture and adhesion. Diapedisis is mediated by a number of molecules including PECAM, PVR, JAMs, and CD99, which have distinct molecular interactions and signaling pathways, but all reside in the LBRC. Much of the field is currently focused on how all these divergent signaling pathways are orchestrated and converge to facilitate the remodeling of junctional molecules to allow for the passage of the migrating leukocyte. Although several of these molecules and pathways appear to be analogous to the mechanisms that regulate vascular permeability on a global level, it is not clear how they are locally regulated during TEM. Transendothelial migration, whether across the endothelial junction or through the cell involves the recruitment of the LBRC and its associated molecules, which are required for efficient TEM. Together, these pathways and the LBRC provide the migrating leukocyte with an appropriate conduit for through which they can reach their target tissue and carry out their essential functions. The degree of crosstalk and interconnection between these pathways will undoubtedly be much more complex than can be appreciated at this time. However, the field is advancing rapidly and every new discovery brings closer novel therapies that can regulate inflammation in a tissue- and disease-specific manner.
References
Ley K, Laudanna C, Cybulsky MI, Nourshargh S (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7(9):678–689
Herter J, Zarbock A (2013) Integrin regulation during leukocyte recruitment. J Immunol 190(9):4451–4457. doi:10.4049/jimmunol.1203179
Schmidt S, Moser M, Sperandio M (2013) The molecular basis of leukocyte recruitment and its deficiencies. Mol Immunol 55(1):49–58. doi:10.1016/j.molimm.2012.11.006
Sperandio M (2006) Selectins and glycosyltransferases in leukocyte rolling in vivo. FEBS J 273(19):4377–4389. doi:10.1111/j.1742-4658.2006.05437.x
Ivetic A (2013) Signals regulating L-selectin-dependent leucocyte adhesion and transmigration. Int J Biochem Cell Biol 45(3):550–555. doi:10.1016/j.biocel.2012.12.023
Zarbock A, Ley K, McEver RP, Hidalgo A (2011) Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood 118(26):6743–6751. doi:10.1182/blood-2011-07-343566
Komarova Y, Malik AB (2010) Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 72:463–493. doi:10.1146/annurev-physiol-021909-135833
Coisne C, Engelhardt B (2011) Tight junctions in brain barriers during central nervous system inflammation. Antioxid Redox Signal 15(5):1285–1303. doi:10.1089/ars.2011.3929
Abbott NJ (2002) Astrocyte-endothelial interactions and blood–brain barrier permeability. J Anat 200(6):629–638
Aird WC (2007) Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res 100(2):158–173. doi:10.1161/01.RES.0000255691.76142.4a
Aird WC (2007) Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res 100(2):174–190. doi:10.1161/01.RES.0000255690.03436.ae
Mammoto A, Mammoto T, Ingber DE (2012) Mechanosensitive mechanisms in transcriptional regulation. J Cell Sci 125(Pt 13):3061–3073. doi:10.1242/jcs.093005
Salameh A, Dhein S (2013) Effects of mechanical forces and stretch on intercellular gap junction coupling. Biochim Biophys Acta 1828(1):147–156. doi:10.1016/j.bbamem.2011.12.030
Tarbell JM (2010) Shear stress and the endothelial transport barrier. Cardiovasc Res 87(2):320–330. doi:10.1093/cvr/cvq146
Abbott NJ, Ronnback L, Hansson E (2006) Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 7(1):41–53. doi:10.1038/nrn1824
Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonniere L, Bernard M, van Horssen J, de Vries HE, Charron F, Prat A (2011) The Hedgehog pathway promotes blood–brain barrier integrity and CNS immune quiescence. Science 334(6063):1727–1731. doi:10.1126/science.1206936
Shaw SK, Bamba PS, Perkins BN, Luscinskas FW (2001) Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J Immunol 167(4):2323–2330
Muller WA (2011) Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol 6:323–344. doi:10.1146/annurev-pathol-011110-130224
Carman CV, Springer TA (2008) Trans-cellular migration: cell–cell contacts get intimate. Curr Opin Cell Biol 20(5):533–540
Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM (1998) Neutrophils emigrate from venules by a transendothelial cell pathway in response to fMLP. J Exp Med 187:903–915
Muller WA (2007) PECAM: Regulating the start of diapedesis. In: Ley K (ed) Adhesion molecules: function and inhibition. Progress in inflammation research. Birkhauser, Basel, pp 201–220
Dejana E, Giampietro C (2012) Vascular endothelial-cadherin and vascular stability. Curr Opin Hematol 19(3):218–223. doi:10.1097/MOH.0b013e3283523e1c
Harris ES, Nelson WJ (2010) VE-cadherin: at the front, center, and sides of endothelial cell organization and function. Curr Opin Cell Biol 22(5):651–658. doi:10.1016/j.ceb.2010.07.006
Vincent PA, Xiao K, Buckley KM, Kowalczyk AP (2004) VE-cadherin: adhesion at arm’s length. Am J Physiol Cell Physiol 286(5):C987–C997. doi:10.1152/ajpcell.00522.2003
Alcaide P, Newton G, Auerbach S, Sehrawat S, Mayadas TN, Golan DE, Yacono P, Vincent P, Kowalczyk A, Luscinskas FW (2008) p120-Catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood 112(7):2770–2779
Schulte D, Kuppers V, Dartsch N, Broermann A, Li H, Zarbock A, Kamenyeva O, Kiefer F, Khandoga A, Massberg S, Vestweber D (2011) Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J 30(20):4157–4170. doi:10.1038/emboj.2011.304
Nanes BA, Chiasson-MacKenzie C, Lowery AM, Ishiyama N, Faundez V, Ikura M, Vincent PA, Kowalczyk AP (2012) p120-catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol 199(2):365–380. doi:10.1083/jcb.201205029
Allingham MJ, van Buul JD, Burridge K (2007) ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol 179(6):4053–4064
Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, Cagna G, Linnepe R, Schulte D, Nottebaum AF, Vestweber D (2011) Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J Exp Med 208(12):2393–2401. doi:10.1084/jem.20110525
Vockel M, Vestweber D (2013) How T cells trigger the dissociation of the endothelial receptor phosphatase VE-PTP from VE-cadherin. Blood 122(14):2512–2522. doi:10.1182/blood-2013-04-499228
Nottebaum AF, Cagna G, Winderlich M, Gamp AC, Linnepe R, Polaschegg C, Filippova K, Lyck R, Engelhardt B, Kamenyeva O, Bixel MG, Butz S, Vestweber D (2008) VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med 205(12):2929–2945
Schnoor M, Lai FP, Zarbock A, Klaver R, Polaschegg C, Schulte D, Weich HA, Oelkers JM, Rottner K, Vestweber D (2011) Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J Exp Med 208(8):1721–1735. doi:10.1084/jem.20101920
von Wedel-Parlow M, Schrot S, Lemmen J, Treeratanapiboon L, Wegener J, Galla HJ (2011) Neutrophils cross the BBB primarily on transcellular pathways: an in vitro study. Brain Res 1367:62–76. doi:10.1016/j.brainres.2010.09.076
Wewer C, Seibt A, Wolburg H, Greune L, Schmidt MA, Berger J, Galla HJ, Quitsch U, Schwerk C, Schroten H, Tenenbaum T (2011) Transcellular migration of neutrophil granulocytes through the blood–cerebrospinal fluid barrier after infection with Streptococcus suis. J Neuroinflammation 8:51. doi:10.1186/1742-2094-8-51
Wolburg H, Wolburg-Buchholz K, Engelhardt B (2005) Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol 109(2):181–190
Proebstl D, Voisin MB, Woodfin A, Whiteford J, D’Acquisto F, Jones GE, Rowe D, Nourshargh S (2012) Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med 209(6):1219–1234. doi:10.1084/jem.20111622
Wang S, Voisin MB, Larbi KY, Dangerfield J, Scheiermann C, Tran M, Maxwell PH, Sorokin L, Nourshargh S (2006) Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med 203(6):1519–1532
Nourshargh S, Hordijk PL, Sixt M (2010) Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol 11(5):366–378. doi:10.1038/nrm2889
Rowe RG, Weiss SJ (2008) Breaching the basement membrane: who, when and how? Trends Cell Biol 18(11):560–574. doi:10.1016/j.tcb.2008.08.007
McEver RP (2002) Selectins: lectins that initiate cell adhesion under flow. Curr Opin Cell Biol 14(5):581–586
Bevilacqua MP (1993) Endothelial-leukocyte adhesion molecules. Annu Rev Immunol 11:767–804. doi:10.1146/annurev.iy.11.040193.004003
Galkina E, Ley K (2007) Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol 27(11):2292–2301. doi:10.1161/ATVBAHA.107.149179
Laudanna C, Kim JY, Constantin G, Butcher E (2002) Rapid leukocyte integrin activation by chemokines. Immunol Rev 186:37–46
Shaw SK, Ma S, Kim MB, Rao RM, Hartman CU, Froio RM, Yang L, Jones T, Liu Y, Nusrat A, Parkos CA, Luscinskas FW (2004) Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med 200(12):1571–1580
Barreiro O, Yanez-Mo M, Sala-Valdes M, Gutierrez-Lopez MD, Ovalle S, Higginbottom A, Monk PN, Cabanas C, Sanchez-Madrid F (2005) Endothelial tetraspanin microdomains regulate leukocyte firm adhesion during extravasation. Blood 105(7):2852–2861. doi:10.1182/blood-2004-09-3606
Yang L, Kowalski JR, Zhan X, Thomas SM, Luscinskas FW (2006) Endothelial cell cortactin phosphorylation by Src contributes to polymorphonuclear leukocyte transmigration in vitro. Circ Res 98(3):394–402
Yang L, Kowalski JR, Yacono P, Bajmoczi M, Shaw SK, Froio RM, Golan DE, Thomas SM, Luscinskas FW (2006) Endothelial cell cortactin coordinates intercellular adhesion molecule-1 clustering and actin cytoskeleton remodeling during polymorphonuclear leukocyte adhesion and transmigration. J Immunol 177(9):6440–6449
Cook-Mills JM (2002) VCAM-1 signals during lymphocyte migration: role of reactive oxygen species. Mol Immunol 39(9):499–508
van Wetering S, van den Berk N, van Buul JD, Mul FP, Lommerse I, Mous R, ten Klooster JP, Zwaginga JJ, Hordijk PL (2003) VCAM-1-mediated Rac signaling controls endothelial cell–cell contacts and leukocyte transmigration. Am J Physiol Cell Physiol 285(2):C343–C352
van Buul JD, Kanters E, Hordijk PL (2007) Endothelial signaling by Ig-like cell adhesion molecules. Arterioscler Thromb Vasc Biol 27(9):1870–1876
Lorenzon P, Vecile E, Nardon E, Ferrero E, Harlan JM, Tedesco F, Dobrina A (1998) Endothelial cell E-and P-selectin and vascular cell adhesion molecule-1 function as signaling receptors. J Cell Biol 142:1381–1391
Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, Furthmayr H, Sanchez-Madrid F (2002) Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol 157(7):1233–1245
Carman CV, Jun CD, Salas A, Springer TA (2003) Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J Immunol 171(11):6135–6144
Carman CV, Springer TA (2004) A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol 167(2):377–388
Mamdouh Z, Mikhailov A, Muller WA (2009) Transcellular migration of leukocytes is mediated by the endothelial lateral border recycling compartment. J Exp Med 206(11):2795–2808
Millan J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ (2006) Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol 8(2):113–123
van Buul JD, Allingham MJ, Samson T, Meller J, Boulter E, Garcia-Mata R, Burridge K (2007) RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol 178(7):1279–1293
Staunton DE, Dustin ML, Springer TA (1989) Functional cloning of ICAM-2, a cell adhesion ligand for LFA-1 homologous to ICAM-1. Nature (London) 339:61–64
de Fougerolles AR, Stacker SA, Schwarting R, Springer TA (1991) Characterization of ICAM-2 and evidence for a third counter-receptor for LFA-1. J Exp Med 174:253–267
McLaughlin F, Hayes BP, Horgan CM, Beesley JE, Campbell CJ, Randi AM (1998) Tumor necrosis factor (TNF)-alpha and interleukin (IL)-1beta down-regulate intercellular adhesion molecule (ICAM)-2 expression on the endothelium. Cell Adhes Commun 6(5):381–400
Springer TA (1994) Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76:301–314
Gerwin N, Gonzalo JA, Lloyd C, Coyle AJ, Reiss Y, Banu N, Wang B, Xu H, Avraham H, Engelhardt B, Springer TA, Gutierrez-Ramos JC (1999) Prolonged eosinophil accumulation in allergic lung interstitium of ICAM-2 deficient mice results in extended hyperresponsiveness. Immunity 10(1):9–19
Huang MT, Larbi KY, Scheiermann C, Woodfin A, Gerwin N, Haskard DO, Nourshargh S (2006) ICAM-2 mediates neutrophil transmigration in vivo: evidence for stimulus specificity and a role in PECAM-1-independent transmigration. Blood 107(12):4721–4727
Hobden JA (2003) Intercellular adhesion molecule-2 (ICAM-2) and Pseudomonas aeruginosa ocular infection. DNA Cell Biol 22(10):649–655. doi:10.1089/104454903770238120
Steiner O, Coisne C, Cecchelli R, Boscacci R, Deutsch U, Engelhardt B, Lyck R (2010) Differential roles for endothelial ICAM-1, ICAM-2, and VCAM-1 in shear-resistant T cell arrest, polarization, and directed crawling on blood–brain barrier endothelium. J Immunol 185(8):4846–4855. doi:10.4049/jimmunol.0903732
Mandell KJ, Parkos CA (2005) The JAM family of proteins. Adv Drug Deliv Rev 57(6):857–867. doi:10.1016/j.addr.2005.01.005
Luscinskas FW, Ma S, Nusrat A, Parkos CA, Shaw SK (2002) The role of endothelial cell lateral junctions during leukocyte trafficking. Immunol Rev 186:57–67
Vaughn DE, Bjorkman PJ (1996) The (Greek) key to structures of neural adhesion molecules. Neuron 16(2):261–273
Ostermann G, Weber KSC, Zernecke A, Schroder A, Weber C (2002) JAM-1 is a ligand of the β2 integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol 3:151–158
Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, Dejana E (1998) Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142:117–127
Johnson-Leger C, Aurrand-Lions M, Beltraminelli N, Fasel N, Imhof BA (2002) Junctional adhesion molecule-2 (JAM-2) promotes lymphocyte transendothelial migration. Blood 100:2479–2486
Shaw SK, Perkins BN, Lim Y-C, Liu Y, Nusrat A, Schnell FJ, Parkos CA, Luscinskas FW (2001) Reduced expression of junctional adhesion molecule and platelet/endothelial cell adhesion molecule-1 (CD31) at human vascular endothelial junctions by cytokines tumor necrosis factor-{alpha} plus interferon-{gamma} does not reduce leukocyte transmigration under flow. Am J Pathol 159(6):2281–2291
Cunningham SA, Rodriguez JM, Arrate MP, Tran TM, Brock TA (2002) JAM2 interacts with alpha 4beta 1. Facilitation by JAM3. J Biol Chem 277(31):27589–27592
Santoso S, Sachs UJ, Kroll H, Linder M, Ruf A, Preissner KT, Chavakis T (2002) The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med 196(5):679–691
Aurrand-Lions M, Lamagna C, Dangerfield JP, Wang S, Herrera P, Nourshargh S, Imhof BA (2005) Junctional adhesion molecule-C regulates the early influx of leukocytes into tissues during inflammation. J Immunol 174(10):6406–6415
Chavakis T, Keiper T, Matz-Westphal R, Hersemeyer K, Sachs UJ, Nawroth PP, Preissner KT, Santoso S (2004) The junctional adhesion molecule-C promotes neutrophil transendothelial migration in vitro and in vivo. J Biol Chem 279:55602–55608
Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE, Meda P, Imhof BA, Nourshargh S (2011) The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol 12(8):761–769. doi:10.1038/ni.2062
Ludwig RJ, Zollner TM, Santoso S, Hardt K, Gille J, Baatz H, Johann PS, Pfeffer J, Radeke HH, Schon MP, Kaufmann R, Boehncke WH, Podda M (2005) Junctional adhesion molecules (JAM)-B and -C contribute to leukocyte extravasation to the skin and mediate cutaneous inflammation. J Invest Dermatol 125(5):969–976. doi:10.1111/j.0022-202X.2005.23912.x
Bradfield PF, Scheiermann C, Nourshargh S, Ody C, Luscinskas FW, Rainger GE, Nash GB, Miljkovic-Licina M, Aurrand-Lions M, Imhof BA (2007) JAM-C regulates unidirectional monocyte transendothelial migration in inflammation. Blood 110(7):2545–2555. doi:10.1182/blood-2007-03-078733
Newman PJ, Berndt MC, Gorski J, White GC 2nd, Lyman S, Paddock C, Muller WA (1990) PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science 247(4947):1219–1222
Wee JL, Jackson DE (2005) The Ig-ITIM superfamily member PECAM-1 regulates the “outside-in” signaling properties of integrin {alpha}IIb{beta}3 in platelets. Blood 106(12):3816–3823
Muller WA, Weigl SA, Deng X, Phillips DM (1993) PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med 178:449–460
Liao F, Ali J, Greene T, Muller WA (1997) Soluble domain 1 of platelet-endothelial cell adhesion molecule (PECAM) is sufficient to block transendothelial migration in vitro and in vivo. J Exp Med 185:1349–1357
Schenkel AR, Mamdouh Z, Muller WA (2004) Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol 5(4):393–400
Mendelsohn CL, Wimmer E, Racaniello VR (1989) Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 56(5):855–865
Sloan KE, Eustace BK, Stewart JK, Zehetmeier C, Torella C, Simeone M, Roy JE, Unger C, Louis DN, Ilag LL, Jay DG (2004) CD155/PVR plays a key role in cell motility during tumor cell invasion and migration. BMC Cancer 4:73. doi:10.1186/1471-2407-4-73
Oda T, Ohka S, Nomoto A (2004) Ligand stimulation of CD155alpha inhibits cell adhesion and enhances cell migration in fibroblasts. Biochem Biophys Res Commun 319(4):1253–1264
Lange R, Peng X, Wimmer E, Lipp M, Bernhardt G (2001) The poliovirus receptor CD155 mediates cell-to-matrix contacts by specifically binding to vitronectin. Virology 285(2):218–227
Mueller S, Wimmer E (2003) Recruitment of nectin-3 to cell–cell junctions through trans-heterophilic interaction with CD155, a vitronectin and poliovirus receptor that localizes to alpha(v)beta3 integrin-containing membrane microdomains. J Biol Chem 278(33):31251–31260
Shibuya K, Shirakawa J, Kameyama T, Honda S, Tahara-Hanaoka S, Miyamoto A, Onodera M, Sumida T, Nakauchi H, Miyoshi H, Shibuya A (2003) CD226 (DNAM-1) is involved in lymphocyte function-associated antigen 1 costimulatory signal for naive T cell differentiation and proliferation. J Exp Med 198(12):1829–1839
Sherrington PD, Scott JL, Jin B, Simmons D, Dorahy DJ, Lloyd J, Brien JH, Aebersold RH, Adamson J, Zuzel M, Burns GF (1997) TLiSA1 (PTA1) activation antigen implicated in T cell differentiation and platelet activation is a member of the immunoglobulin superfamily exhibiting distinctive regulation of expression. J Biol Chem 272(35):21735–21744
Kojima H, Kanada H, Shimizu S, Kasama E, Shibuya K, Nakauchi H, Nagasawa T, Shibuya A (2003) CD226 mediates platelet and megakaryocytic cell adhesion to vascular endothelial cells. J Biol Chem 278(38):36748–36753
Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, Kitamura T, Nicholl J, Sutherland GR, Lanier LL, Phillips JH (1996) DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity 4(6):573–581
Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M (2004) Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J Immunol 172(7):3994–3998
Sullivan DP, Seidman MA, Muller WA (2013) Poliovirus receptor (CD155) regulates a step in transendothelial migration between PECAM and CD99. Am J Pathol 182(3):1031–1042. doi:10.1016/j.ajpath.2012.11.037
Manes TD, Pober JS (2011) Identification of endothelial cell junctional proteins and lymphocyte receptors involved in transendothelial migration of human effector memory CD4+ T cells. J Immunol 186(3):1763–1768. doi:10.4049/jimmunol.1002835
Reymond N, Imbert AM, Devilard E, Fabre S, Chabannon C, Xerri L, Farnarier C, Cantoni C, Bottino C, Moretta A, Dubreuil P, Lopez M (2004) DNAM-1 and PVR regulate monocyte migration through endothelial junctions. J Exp Med 199(10):1331–1341
Bernhardt G, Bibb JA, Bradley J, Wimmer E (1994) Molecular characterization of the cellular receptor for poliovirus. Virology 199(1):105–113
Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, Cantoni C, Grassi J, Marcenaro S, Reymond N, Vitale M, Moretta L, Lopez M, Moretta A (2003) Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med 198(4):557–567
Freistadt MS, Eberle KE (1997) Physical association between CD155 and CD44 in human monocytes. Mol Immunol 34(18):1247–1257
Koike S, Horie H, Ise I, Okitsu A, Yoshida M, Iizuka N, Takeuchi K, Takegami T, Nomoto A (1990) The poliovirus receptor protein is produced both as membrane-bound and secreted forms. EMBO J 9(10):3217–3224
Ohka S, Ohno H, Tohyama K, Nomoto A (2001) Basolateral sorting of human poliovirus receptor alpha involves an interaction with the mu1B subunit of the clathrin adaptor complex in polarized epithelial cells. Biochem Biophys Res Commun 287(4):941–948
Jia W, Liu XS, Zhu Y, Li Q, Han WN, Zhang Y, Zhang JS, Yang K, Zhang XH, Jin BQ (2000) Preparation and characterization of mabs against different epitopes of CD226 (PTA1). Hybridoma 19(6):489–494
Chen L, Xie X, Zhang X, Jia W, Jian J, Song C, Jin B (2003) The expression, regulation and adhesion function of a novel CD molecule, CD226, on human endothelial cells. Life Sci 73(18):2373–2382
Gelin C, Aubrit F, Phalipon A, Raynal B, Cole S, Kaczorek M, Bernard A (1989) The E2 antigen, a 32 kd glycoprotein involved in T-cell adhesion processes, is the MIC2 gene product. EMBO J 8:3253–3259
Suh YH, Shin YK, Kook MC, Oh KI, Park WS, Kim SH, Lee IS, Park HJ, Huh TL, Park SH (2003) Cloning, genomic organization, alternative transcripts and expression analysis of CD99L2, a novel paralog of human CD99, and identification of evolutionary conserved motifs. Gene 307:63–76
Bixel MG, Petri B, Khandoga AG, Khandoga A, Wolburg-Buchholz K, Wolburg H, Marz S, Krombach F, Vestweber D (2007) A CD99-related antigen on endothelial cells mediates neutrophil, but not lymphocyte extravasation in vivo. Blood 109:5327–5336
Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA (2002) CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol 3(2):143–150
Schenkel AR, Dufour EM, Chew TW, Sorg E, Muller WA (2007) The murine CD99-related molecule CD99-like 2 (CD99L2) is an adhesion molecule involved in the inflammatory response. Cell Commun Adhes 14(5):227–237
Seelige R, Natsch C, Marz S, Jing D, Frye M, Butz S, Vestweber D (2013) Cutting edge: endothelial-specific gene ablation of CD99L2 impairs leukocyte extravasation in vivo. J Immunol 190(3):892–896. doi:10.4049/jimmunol.1202721
Nam G, Lee YK, Lee HY, Ma MJ, Araki M, Araki K, Lee S, Lee IS, Choi EY (2013) Interaction of CD99 with its paralog CD99L2 positively regulates CD99L2 trafficking to cell surfaces. J Immunol 19(11):5730–5742. doi:10.4049/jimmunol.1203062
Lou O, Alcaide P, Luscinskas FW, Muller WA (2007) CD99 is a key mediator of the transendothelial migration of neutrophils. J Immunol 178:1136–1143
Bixel MG, Li H, Petri B, Khandoga AG, Khandoga A, Zarbock A, Wolburg-Buchholz K, Wolburg H, Sorokin L, Zeuschner D, Maerz S, Butz S, Krombach F, Vestweber D (2010) CD99 and CD99L2 act at the same site as, but independently of, PECAM-1 during leukocyte diapedesis. Blood 116(7):1172–1184. doi:10.1182/blood-2009-12-256388
Worthylake RA, Burridge K (2001) Leukocyte transendothelial migration: orchestrating the underlying molecular machinery. Curr Opin Cell Biol 13(5):569–577
Voisin MB, Nourshargh S (2013) Neutrophil transmigration: emergence of an adhesive cascade within venular walls. J Innate Immun 5(4):336–347. doi:10.1159/000346659
Schenkel AR, Chew TW, Muller WA (2004) Platelet endothelial cell adhesion molecule deficiency or blockade significantly reduces leukocyte emigration in a majority of mouse strains. J Immunol 173(10):6403–6408
Liao F, Huynh HK, Eiroa A, Greene T, Polizzi E, Muller WA (1995) Migration of monocytes across endothelium and passage through extracellular matrix involve separate molecular domains of PECAM-1. J Exp Med 182:1337–1343
Woodfin A, Voisin MB, Imhof BA, Dejana E, Engelhardt B, Nourshargh S (2009) Endothelial cell activation leads to neutrophil transmigration as supported by the sequential roles of ICAM-2, JAM-A and PECAM-1. Blood 113(24):6246–6257
Mamdouh Z, Chen X, Pierini LM, Maxfield FR, Muller WA (2003) Targeted recycling of PECAM from endothelial cell surface-connected compartments during diapedesis. Nature 421:748–753
Alcaide P, Auerbach S, Luscinskas FW (2009) Neutrophil recruitment under shear flow: it’s all about endothelial cell rings and gaps. Microcirculation 16(1):43–57. doi:10.1080/10739680802273892
Allport JR, Muller WA, Luscinskas FW (2000) Monocytes induce reversible focal changes in vascular endothelial cadherin complex during transendothelial migration under flow. J Cell Biol 148(1):203–216
Jin S, Umemoto E, Tanaka T, Shimomura Y, Tohya K, Kunizawa K, Yang BG, Jang MH, Hirata T, Miyasaka M (2008) Nepmucin/CLM-9, an Ig domain-containing sialomucin in vascular endothelial cells, promotes lymphocyte transendothelial migration in vitro. FEBS Lett 582(20):3018–3024
Mamdouh Z, Kreitzer GE, Muller WA (2008) Leukocyte transmigration requires kinesin-mediated microtubule-dependent membrane trafficking from the lateral border recycling compartment. J Exp Med 205(4):951–966
Bird IN, Spragg JH, Ager AH, Matthews N (1993) Studies of lymphocyte transendothelial migration: analysis of migrated cell phenotypes with regard to CD31 (PECAM-1), CD45RA and CD45RO. Immunology 80:553–560
Muller WA (2001) Migration of leukocytes across endothelial junctions: some concepts and controversies. Microcirculation 8:181–193
Dvorak AM, Kohn S, Morgan ES, Fox P, Nagy JA, Dvorak HF (1996) The vesiculo-vacuolar organelle (VVO): a distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. J Leukoc Biol 59(1):100–115
Feng D, Nagy JA, Hipp J, Dvorak HF, Dvorak AM (1996) Vesiculo-vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin. J Exp Med 183:1981–1986
Rothberg KG, Heuser JE, Donzell WC, Ying Y-S, Glenney JR, Anderson RGW (1992) Caveolin, a protein component of caveolae membrane coats. Cell 68:673–682
Dvorak AM, Feng D (2001) The vesiculo-vacuolar organelle (VVO). A new endothelial cell permeability organelle. J Histochem Cytochem 49(4):419–432
Florey O, Durgan J, Muller W (2010) Phosphorylation of leukocyte PECAM and its association with detergent-resistant membranes regulate transendothelial migration. J Immunol 185(3):1878–1886
Sardjono CT, Harbour SN, Yip JC, Paddock C, Tridandapani S, Newman PJ, Jackson DE (2006) Palmitoylation at Cys595 is essential for PECAM-1 localisation into membrane microdomains and for efficient PECAM-1-mediated cytoprotection. Thromb Haemost 96(6):756–766
Sallese M, Giannotta M, Luini A (2009) Coordination of the secretory compartments via inter-organelle signalling. Semin Cell Dev Biol 20(7):801–809. doi:10.1016/j.semcdb.2009.04.004
Teasdale RD, Jackson MR (1996) Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the golgi apparatus. Annu Rev Cell Dev Biol 12:27–54
Bonifacino JS, Traub LM (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 72:395–447
Dasgupta B, Dufour E, Mamdouh Z, Muller W (2009) A novel and critical role for tyrosine 663 in PECAM trafficking and transendothelial migration. J Immunol 182(8):5041–5051
Dasgupta B, Muller WA (2008) Endothelial Src kinase regulates membrane recycling from the lateral border recycling compartment during leukocyte transendothelial migration. Eur J Immunol 38(12):3499–3507
Coyne CB, Kim KS, Bergelson JM (2007) Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. EMBO J 26(17):4016–4028. doi:10.1038/sj.emboj.7601831
Sage PT, Carman CV (2009) Settings and mechanisms for trans-cellular diapedesis. Front Biosci 14:5066–5083
Wittchen ES (2009) Endothelial signaling in paracellular and transcellular leukocyte transmigration. Front Biosci (Landmark Ed) 14:2522–2545
Engelhardt B (2008) Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J Neurol Sci 274(1–2):23–26. doi:10.1016/j.jns.2008.05.019
Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, Dvorak HF, Dvorak AM, Springer TA (2007) Transcellular diapedesis is initiated by invasive podosomes. Immunity 26(6):784–797
Rizo J, Sudhof TC (2012) The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices—guilty as charged? Annu Rev Cell Dev Biol 28:279–308. doi:10.1146/annurev-cellbio-101011-155818
Jahn R, Fasshauer D (2012) Molecular machines governing exocytosis of synaptic vesicles. Nature 490(7419):201–207. doi:10.1038/nature11320
Goddard LM, Iruela-Arispe ML (2013) Cellular and molecular regulation of vascular permeability. Thromb Haemost 109(3):407–415. doi:10.1160/TH12-09-0678
Tilghman RW, Hoover RL (2002) The Src-cortactin pathway is required for clustering of E-selectin and ICAM-1 in endothelial cells. FASEB J 16(10):1257–1259. doi:10.1096/fj.01-0969fje
Durieu-Trautmann O, Chaverot N, Cazaubon S, Strosberg AD, Couraud PO (1994) Intercellular adhesion molecule 1 activation induces tyrosine phosphorylation of the cytoskeleton-associated protein cortactin in brain microvessel endothelial cells. J Biol Chem 269(17):12536–12540
Timmerman I, Hoogenboezem M, Bennett AM, Geerts D, Hordijk PL, van Buul JD (2012) The tyrosine phosphatase SHP2 regulates recovery of endothelial adherens junctions through control of beta-catenin phosphorylation. Mol Biol Cell 23(21):4212–4225. doi:10.1091/mbc.E12-01-0038
Couty JP, Rampon C, Leveque M, Laran-Chich MP, Bourdoulous S, Greenwood J, Couraud PO (2007) PECAM-1 engagement counteracts ICAM-1-induced signaling in brain vascular endothelial cells. J Neurochem 103(2):793–801
Liu G, Place AT, Chen Z, Brovkovych VM, Vogel SM, Muller WA, Skidgel RA, Malik AB, Minshall RD (2012) ICAM-1-activated Src and eNOS signaling increase endothelial cell surface PECAM-1 adhesivity and neutrophil transmigration. Blood 120(9):1942–1952. doi:10.1182/blood-2011-12-397430
Ukropec JA, Hollinger MK, Salva SM, Woolkalis MJ (2000) SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. J Biol Chem 275(8):5983–5986
Gratzinger D, Barreuther M, Madri JA (2003) Platelet-endothelial cell adhesion molecule-1 modulates endothelial migration through its immunoreceptor tyrosine-based inhibitory motif. Biochem Biophys Res Commun 301(1):243–249
Masuda M, Osawa M, Shigematsu H, Harada N, Fujiwara K (1997) Platelet endothelial cell adhesion molecule-1 is a major SH-PTP2 binding protein in vascular endothelial cells. FEBS Lett 408(3):331–336
Martinelli R, Gegg M, Longbottom R, Adamson P, Turowski P, Greenwood J (2009) ICAM-1-mediated endothelial nitric oxide synthase activation via calcium and AMP-activated protein kinase is required for transendothelial lymphocyte migration. Mol Biol Cell 20(3):995–1005. doi:10.1091/mbc.E08-06-0636
van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW (2000) Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res 87(4):335–340
Saito H, Minamiya Y, Kitamura M, Saito S, Enomoto K, Terada K, Ogawa J (1998) Endothelial myosin light chain kinase regulates neutrophil migration across human umbilical vein endothelial cell monolayer. J Immunol 161:1533–1540
Kielbassa-Schnepp K, Strey A, Janning A, Missiaen L, Nilius B, Gerke V (2001) Endothelial intracellular Ca2+ release following monocyte adhesion is required for the transendothelial migration of monocytes. Cell Calcium 30(1):29–40. doi:10.1054/ceca.2001.0210
Cook-Mills JM, Johnson JD, Deem TL, Ochi A, Wang L, Zheng Y (2004) Calcium mobilization and Rac1 activation are required for VCAM-1 (vascular cell adhesion molecule-1) stimulation of NADPH oxidase activity. Biochem J 378(Pt 2):539–547
Su WH, Chen HI, Huang JP, Jen CJ (2000) Endothelial [Ca(2+)](i) signaling during transmigration of polymorphonuclear leukocytes. Blood 96(12):3816–3822
Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC (1993) Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J Cell Biol 120:1371–1380
Moll TE, Dejana E, Vestweber D (1998) In vitro degradation of endothelial catenins by a neutrophil protease. J Cell Biol 140:403–407
Gotsch U, Borges E, Bosse R, Boggemeyer E, Simon M, Mossmann H, Vestweber D (1997) VE-cadherin antibody accelerates neutrophil recruitment in vivo. J Cell Sci 110:583–588
Xiao K, Garner J, Buckley KM, Vincent PA, Chiasson CM, Dejana E, Faundez V, Kowalczyk AP (2005) p120-Catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol Biol Cell 16(11):5141–5151
Xiao K, Allison DF, Buckley KM, Kottke MD, Vincent PA, Faundez V, Kowalczyk AP (2003) Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol 163(3):535–545. doi:10.1083/jcb.200306001
Acknowledgments
This work was supported by NIH R01 HL046849 and R37 HL064774 (to W.A.M.) and NIH F32 to AI084454 (to D.P.S.) We thank Ryan Winger for thoughtful discussions regarding this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on New paradigms in leukocyte trafficking, lessons for therapeutics - Guest Editors: F. W. Luscinskas and B. A. Imhof
Rights and permissions
About this article
Cite this article
Sullivan, D.P., Muller, W.A. Neutrophil and monocyte recruitment by PECAM, CD99, and other molecules via the LBRC. Semin Immunopathol 36, 193–209 (2014). https://doi.org/10.1007/s00281-013-0412-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-013-0412-6