
Abstract
Endoplasmic reticulum (ER) stress and consequent unfolded protein response (UPR) are involved in a diverse range of pathologies including ischemic diseases, neurodegenerative disorders, and metabolic diseases, such as diabetes mellitus. The UPR is also triggered by various environmental factors; e.g., pollutants, infectious pathogens, therapeutic drugs, alcohol, physical stress, and malnutrition. This review summarizes current knowledge on environmental factors that induce ER stress and describes how the UPR is linked to particular pathological states after exposure to environmental triggers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The endoplasmic reticulum (ER) provides a unique environment for appropriate protein folding, assembly, and disulfide bond formation, leading to production of mature proteins. Various pathophysiological insults lead to accumulation of unfolded proteins in the ER, namely ER stress. Those include hypoxia, nutrient deprivation, altered redox balance, changes in calcium homeostasis, failure of post-translational modifications, and increase in general protein synthesis [1]. In response to ER stress, cells adapt themselves to the stress condition via the unfolded protein response (UPR); that is, attenuation of general translation, induction of ER chaperones, and reinforcement of ER-associated degradation (ERAD) to eliminate immature proteins. When the stress is beyond the capacity of the pro-survival capabilities of the UPR, however, cells undergo apoptosis through the pro-apoptotic component of the UPR. Consistent with this, ER stress and the UPR are likely to be involved in a diverse range of pathological situations including ischemic injury, viral infection, neurodegenerative disorders, drug-induced tissue injury and metabolic diseases such as diabetes mellitus which are associated with cellular stress [1, 2].
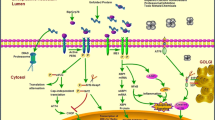
Three major transducers are present on the membrane of the ER for sensing ER stress. These are RNA-dependent protein kinase-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1). Activation of PERK leads to phosphorylation of eukaryotic translation initiation factor 2α (eIF2α), which causes general inhibition of protein synthesis. In response to ER stress, p90ATF6 transits to the Golgi where it is cleaved by the proteases S1P and S2P, yielding a free cytoplasmic domain (p50ATF6 or ATF6f) that functions as a transcription factor. Similarly, activated IRE1α, the ubiquitously expressed isoform of IRE1, catalyzes removal of a small intron from the mRNA encoding X-box binding protein 1 (XBP1). This splicing event creates a translational frameshift in XBP1 mRNA to produce an active transcription factor. Active p50ATF6 and XBP1 subsequently bind to the ER stress response element (ERSE) and the UPR element (UPRE) associated with many target genes, leading to expression of proteins that assist ER-associated folding such as ER chaperone 78-kDa glucose-regulated protein (GRP78) and ERAD factors that promote protein removal. These pathways are generally regarded as pro-survival UPR pathways [3] (Fig. 1).

The UPR elicited by environmental factors. Environmental factors such as chemical pollutants, therapeutic drugs, infectious pathogens, alcohol, physical stress, and malnutrition cause ER stress. ER stress is sensed by three major transducers in the ER membrane; PERK, ATF6, and IRE1. Activation of PERK leads to phosphorylation of eIF2α that causes general translational inhibition and consequent attenuation of ER stress. It also causes induction of ATF4, activation of AARE, and consequent expression of target genes including CHOP. In response to ER stress, p90ATF6 is transmitted to the Golgi where it is cleaved by the proteases S1P and S2P, yielding a free cytoplasmic domain p50ATF6 that functions as a transcription factor. Similarly, activated IRE1 (endoribonuclease activity) catalyzes removal of a small intron from XBP1 mRNA, resulting in generation of an active transcription factor. p50ATF6 and XBP1 subsequently bind to ERSE and UPRE, leading to expression of target genes including ER chaperones and ERAD factors. These pathways attenuate ER stress by promoting protein folding and facilitating degradation of unfolded proteins. On the other hand, ER stress also triggers cleavage of procaspase-12 (or procaspase-4 in humans) localized at the ER membrane, leading cells to undergo apoptosis. The IRE1-TRAF2 interaction also allows for recruitment of ASK1 and activation of JNK, leading to apoptotic cell death
During the UPR, however, death signals may also be transduced [4]. For example, expression of pro-apoptotic CCAAT/enhancer-binding protein-homologous protein (CHOP) (also called growth arrest and DNA damage-inducible protein 153) is generated by binding of ATF4 to the amino acid response element (AARE) that is activated by the PERK–eIF2α pathway. ER stress also activates caspase-12 (or caspase-4 in humans), which is localized at the ER membrane through an interaction with IRE1 and tumor necrosis factor receptor-associated factor 2 (TRAF2), leading cells to undergo apoptosis. The IRE1–TRAF2 interaction also allows for recruitment and activation of apoptosis signal-regulating kinase 1 (ASK1) and downstream c-Jun N-terminal kinase (JNK), both of which are involved in a variety of pro-apoptotic signaling [4] (Fig. 1).
A common feature of many of the aforementioned diseases is that they have a significant environmental contribution in their pathogenesis. In this review, I therefore summarize current knowledge on environmental factors that trigger the UPR and might then contribute to disease. Those include pollutants (toxic metals, cigarette smoke, paraquat, silica nanoparticle, formaldehyde, bis(tri-n-butyltin)oxide, nitro-polycyclic aromatic hydrocarbon (nitro-PAH)), infectious pathogens (bacteria, virus, and prion), therapeutic drugs (anti-Inflammatory drugs, immunosuppressants, anti-cancer agents, antibiotics, human immunodeficiency virus (HIV) protease inhibitors, psychotropic drugs), alcohol, physical stress [acidic stress, thermal stress, ultraviolet (UV) light and X-ray, hypoxia] and malnutrition (glucose, amino acids, vitamins). This review also describes how the UPR is linked to particular pathologies under exposure to individual environmental factors. The current knowledge described in this article is summarized in Table 1.
Pollutants
Toxic metals
Toxic metals, such as cadmium, mercury and lead, pose serious risks to human health. Exposure to these metals causes damage of a variety of organs. One of the most typical examples is nephrotoxicity. Previous studies have suggested that the toxic effect of cadmium on renal tubules is caused by several mechanisms. These include structural alteration of junctional complexes (disruption of tight and gap junctions) and cellular death caused by oxidative stress [5, 6]. In addition to these mechanisms, previous reports have provided evidence for a pathological role of ER stress in cadmium-induced apoptosis of renal tubular cells [7, 8]. Specifically, exposure of cultured tubular cells to cadmium triggered induction of the PERK–eIF2α, ATF6 and IRE1 pathways [7]. The induction of ER stress was also observed by in vivo administration of cadmium into mice, preferentially in the kidney and liver [9]. We identified that the IRE1 pathway promoted a pro-apoptotic UPR via phosphorylation of JNK. In contrast, the PERK–eIF2α pathway was anti-apoptotic and counteracted the pro-apoptotic process [7].
ER stress is induced not only by cadmium but also by various other toxic metals including mercury, lead, copper, nickel, arsenic, and silver [7, 9–13]. This suggests that ER stress may be a common consequence of metal-induced cell injury. Of note, induction of ER stress by toxic metals is not specific to the kidney but is also observed in other tissues and cell types including placenta, hepatomas, gliomas, astrocytes, and endothelial cells [9, 10, 14–16].
Previous studies have shown an involvement of reactive oxygen species (ROS) in cadmium-induced cell injury [17, 18]. We found that cadmium-induced ER stress was inhibited by antioxidants. In contrast, suppression of ER stress did not attenuate cadmium-triggered oxidative stress, suggesting that ER stress is downstream of the oxidative stress [19]. In this study, we also observed that O2 ·− was selectively involved in cadmium-triggered, ER stress-mediated apoptosis through activation of the ATF6 and IRE1 pathways. In addition, phosphorylation of JNK by O2 ·− was particularly important for the induction of apoptosis [19]. In addition, we recently found that inhibition of mammalian target of rapamycin complex 1 (mTORC1) attenuated cadmium-induced apoptosis in renal tubular cells. The activation of mTORC1 by ROS contributed to induction of a selective UPR, specifically the IRE1α pathway. Inhibition of mTORC1 caused suppression of JNK phosphorylation without affecting other UPR pathways in cadmium-exposed cells (our unpublished data). These results suggest mTORC1 mediates cadmium-induced apoptosis through selective induction of IRE1α signaling.
Regarding the mechanisms underlying induction of the UPR by metals, a direct interaction between ER chaperones and metals may be important. Chaperones in the ER play pivotal roles in folding, assembly and post-translational modification of secretory proteins and also contribute to the recycling, refolding and degradation of misfolded proteins. Dysfunction of ER chaperones, therefore, causes protein folding disorders. Recent studies have suggested that the function of ER chaperones is disturbed by their physical interaction with metals, which may cause the UPR and ER stress-related disorders. For example, the “master chaperone” GRP78 is located at the hub of signaling pathways in the complex chaperone network, and toxic metals including lead, copper and aluminum have the potential to bind to GRP78 and impair its chaperone function, as recently reviewed by Tiffany-Castiglioni and Qian [20]. Alternatively, metal-induced ROS may also be involved in the induction of ER stress. This issue is discussed in detail later.
Cigarette smoke
Chronic obstructive pulmonary disease (COPD) is one of the most common causes of death in the world, and the mortality rate continues to increase. Cigarette smoke has been regarded as the pivotal risk factor for COPD characterized by injury of cellular components in the respiratory tract. A previous report has suggested that, in mice and humans with COPD, ER stress is induced in the lungs [21]. We showed that ER stress plays a crucial role in cigarette smoke-triggered apoptosis of bronchial epithelial cells and that CHOP is responsible for the induction of apoptosis [22]. We also observed that, as in the case of cadmium-induced ER stress, cigarette smoke extract causes oxidative stress, which is responsible for the induction of ER stress and consequent expression of CHOP [22].
Cigarette smoke contains thousands of chemicals in the aqueous, gas and tar phases [23]. In the gas phase, the smoke comprises ROS and reactive nitrogen species (RNS) including O2 ·− and NO. The tar phase of cigarette smoke also contains organic radicals (long-lived semiquinone radicals) that react with molecular oxygen to form O2 ·−, H2O2 and OH· [23]. These ROS/RNS may directly affect cellular function. On the other hand, cigarette smoke triggers generation of ROS in mammalian cells and thereby induces the UPR [22]. We tested several major constituents of cigarette smoke for the potential to induce ER stress and found that nicotine, heavy metals and PAHs were not the responsible factors [24].
Acrolein is a major component of cigarette smoke [25]. Some reports have suggested that acrolein may induce ER stress in cultured endothelial cells and pulmonary cells in vitro and in vivo [26, 27]. We tested an effect of this substance on the expression of ER stress markers in bronchial epithelial cells. Northern blot analysis showed that acrolein (10 μM) induced expression of CHOP, and it was associated with significant activation of the CHOP gene promoter [24]. Of note, based on previous data reported by Smith et al. [28], the concentration of acrolein in the cigarette smoke extract was approximately 5–10 μM.
Paraquat
Paraquat is an organic heterocyclic herbicide widely used throughout the world. Epidemiological and neuropathological studies have shown that chronic exposure to paraquat increases the risk of Parkinson's disease, a protein folding disorder [29]. Parkinson’s disease is characterized by loss of dopaminergic neurons, and paraquat is known to cause dopaminergic cell death [29]. Previous studies have shown that paraquat triggers the PERK and IRE1 pathways [30, 31], and a small co-chaperone protein p23 is cleaved during paraquat-induced cell death [30]. Paraquat also inhibits proteasomal activity that may further enhance accumulation of misfolded proteins, resulting in reinforcement of ER stress [30]. It is worth noting that paraquat exposure causes oxidative stress [32]. The induction of ER stress by paraquat may occur, at least in part, via generation of ROS. Why dopaminergic neurons are uniquely susceptible to paraquat is unknown.
Silica nanoparticles
Silica nanoparticles are used in various fields (e.g., chemical mechanical polishing, and medical applications); whereas, they are possibly toxic to humans [33]. Induction of oxidative stress, membrane damage, and disturbed calcium homeostasis are possible mechanisms underlying this cellular toxicity. Because oxidative stress and disturbed calcium homeostasis are triggers of ER stress, silica nanoparticles may induce the UPR. Indeed, a recent report provided evidence for induction of ER stress by silica nanoparticles [34]. Using hepatocytes and fibroblasts, the authors showed that silica nanoparticles induced expression of GRP78 and splicing of XBP1 mRNA, in parallel with generation of ROS. This result raises the possibility that silica nanoparticles cause cell injury via ROS-dependent induction of ER stress.
Formaldehyde
Formaldehyde is a common environmental pollutant found in cigarette smoke, household products, exhaust gas, and many other medical and industrial products. It is a major pollutant responsible for “sick house syndrome.” Formaldehyde possesses neurotoxicity and, after long exposures, it may cause neurodegenerative disorders. Using PC12 neuroblastoma cells, a recent report showed that GRP78 and CHOP are induced by formaldehyde and that it is associated with cleavage of procaspase-12 [35]. Although supportive evidence is limited, toxicity of formaldehyde may be, in part, ascribed to induction of ER stress.
Bis(tri-n-butyltin)oxide
Bis(tri-n-butyltin)oxide (TBTO) belongs to the class of organotins and has been widely used as biocide in wood preservatives and antifouling paints. In rodents exposed to TBTO, immunosuppression is the most prominent toxicity. It causes a reduction in thymus size and the number of thymocytes through induction of apoptosis. The mechanisms underlying the induction of apoptosis are not fully understood, but recent reports have implicated the involvement of ER stress [36, 37]. Microarray analysis and quantitative RT-PCR has revealed that TBTO up-regulates ER stress markers such as Herpud1, Atf3, Ddit3, Edem1, Hspa5 (Grp78), Xbp1, and Eif2α in rat thymus [37] and ATF3, DDIT3 (CHOP), NOXA (PMAIP1), ERO1LB, and HERPUD1 in human T lymphocytes [36]. It is worthwhile noting that TBTO also induces expression of genes involved in the oxidative stress response.
Nitro-polycyclic aromatic hydrocarbon
Epidemiologic studies provide evidence for a link between exposure to air pollution and vascular events including progression of atherosclerotic plaques, altered coagulation and venous thrombosis. In particular, exposure to environmental contaminants such as polycyclic aromatic hydrocarbons (PAHs) and nitro-PAHs increase the incidence of cardiovascular diseases. 1-nitropyrene (1-NP) is one of the most abundant nitro-PAHs in diesel exhaust and a major substance responsible for the mutagenicity of diesel exhaust. Using human umbilical vein endothelial cells, a recent report showed that low levels of 1-NP-induced DNA damage, generation of ROS and induction of GRP78 [38]. Inhibition of nitroreductive metabolism attenuated these molecular events. These results indicate that 1-NP may cause DNA damage in vascular endothelial cells through ROS-mediated induction of the UPR.
Infectious pathogens
Bacteria
Lipopolysaccharide
Lipopolysaccharides (LPSs), also known as lipoglycans, are large molecules that consist of lipid and polysaccharide. They are found in the outer membrane of Gram-negative bacteria, act as endotoxins and elicit strong immune responses in animals. Previous reports have shown that systemic administration of LPS induces ER stress in the lung, which is responsible for endotoxin-induced lung injury [39, 40]. However, the induction of ER stress by LPS is not restricted to the lung but also observed in other organs. We found that after systemic administration with LPS in mice, expression of GRP78 is induced in the liver, kidney, spleen, and heart as well as lung [41]. Only in the brain, induction of GRP78 was not detectable. These results suggest that infection with Gram-negative bacteria induce ER stress in various organs. The molecular mechanisms involved in the induction of ER stress by LPS have not been fully elucidated, but protein overload in the ER (overproduction of inflammatory proteins), production of NO and/or generation of ROS may be possible mechanisms [42, 43].
Several previous reports have shown that ER stress has the potential to induce activation of nuclear factor-κB (NF-κB), the key regulator of immune and inflammatory responses. Various ER stress inducers trigger DNA binding of NF-κB as well as NF-κB-dependent gene expression, leading to induction of inflammation [44, 45]. The induction of ER stress may, in part, contribute to activation of NF-κB by LPS. However, the role of ER stress in inflammation is not so simple as previously proposed. We found that when ER stress is present, activation of NF-κB by inflammatory stimuli is blunted. For example, expression of NF-κB-dependent genes by cytokines is abrogated by pretreatment with ER stress inducers in vitro [46–48]. Consistent with these results, preconditioning with ER stress inducers attenuates LPS-induced endotoxic lethality and collagen arthritis in mice [49]. Although ER stress triggers activation of NF-κB in the early phase, the consequent UPR has the potential to inhibit NF-κB activation in later phases. Several molecular mechanisms are postulated for the negative regulation of NF-κB by the UPR [50, 51]. Therefore, ER stress may play a role not only in the activation of NF-κB but also in the negative feedback loop of NF-κB activity in LPS-exposed cells.
Shiga toxins
Shiga toxin-producing bacteria cause outbreaks of bloody diarrhea, which may progress to life-threatening systemic complications. Shiga toxins, the main virulence factors, activate multiple stress signaling pathway including the UPR [52]. Currently, the mechanisms involved in the induction of ER stress induced by Shiga toxin are not well understood, but a previous report has suggested that its enzymatic activity is required for optimal activation of PERK and ATF6, but not IRE1 [53].
Subtilase cytotoxin (SubAB), the prototype of a distinct AB5 cytotoxin family, was discovered in a highly virulent Shiga toxigenic Escherichia coli strain responsible for an outbreak of hemolytic uremic syndrome [54, 55]. SubAB is a serine protease that selectively cleaves the master ER chaperone GRP78 [56]. As a result of this activity, SubAB is one of the most potent bacterial toxins that selectively induces ER stress, leading to activation of the three major branches of the UPR [57]. Indeed, systemic administration with SubAB triggers the UPR in mice [49]. We found that SubAB induced activation of NF-κB in the early phase [58], whereas subsequent activation of the UPR inhibited NF-κB in the later phase [48, 49]. The ATF6 pathway may play a pivotal role in the biphasic, bidirectional regulation of NF-κB by SubAB [48, 58].
Viruses
Recent studies have suggested the importance of the ER stress response in viral infection. As a processing plant for the folding and post-translational modification of proteins, the ER is an essential organelle for viral replication and maturation [59]. In the course of productive infection, a large amount of viral proteins are synthesized in infected cells, and the consequent accumulation of unfolded or misfolded proteins activates the UPR. The UPR may also be elicited directly via interaction of viruses (viral proteins) and GRP78 (and/or other endogenous UPR constituents) [59]. A number of previous reports have shown that infection by a wide range of viruses including hepatitis B virus, hepatitis C virus, hepatitis D virus, flavivirus, Borna disease virus, murine leukemia virus and Moloney murine leukemia virus induce ER stress [60]. The UPR triggered by viral infection may either protect cells from apoptosis or facilitate the cells to undergo apoptosis. The latter is involved in hepatic injury, carcinogenesis and neurodegeneration caused by viral infection [60].
The UPR induced by viruses, in turn, modulates replication of viruses positively or negatively. Several studies have suggested a connection between the UPR and viral replication. Among the three major UPR branches, the PERK pathway plays a role in limiting viral replication. Activation of PERK leads to phosphorylation of eIF2α, resulting in shutoff of protein synthesis and inhibition of viral replication [59].
Prions
Prion diseases are characterized by the accumulation of misfolded prion protein PrP(Sc) and consequent neuronal death by apoptosis. Hetz et al. reported that nanomolar concentrations of purified PrP(Sc) from mouse scrapie brain-induced apoptosis of neuroblastoma cells. This PrP(Sc) toxicity was associated with an increase in intracellular calcium released from the ER and induction of ER stress markers [61]. Activation of caspase-12 was detected in cells treated with PrP(Sc), and cellular death was inhibited by functional knockdown of caspase-12. In scrapie-infected mice, a correlation between caspase-12 activation and neuronal loss was observed in histological and biochemical analyses. The extent of prion replication was closely correlated with up-regulation of ER stress markers. Similar results were also observed in humans with sporadic and variant Creutzfeldt-Jakob disease [61]. These results suggest a role of the ER stress-related, caspase-12-dependent pathway in the development of neurodegenerative prion diseases.
Drugs
Nonsteroidal anti-inflammatory drugs
A previous study has shown that several non-steroidal anti-inflammatory drugs (NSAIDs) including indomethacin, diclofenac, ibuprofen and celecoxib induce ER stress in gastric mucosal cells [62]. Exposure of cells to indomethacin induced expression of GRP78 and CHOP, which was associated with induction or activation of ATF6, ATF4, and XBP1. NSAIDs other than indomethacin (e.g., diclofenac, ibuprofen, and celecoxib) also induced CHOP. Indomethacin causes apoptosis of gastric mucosal cells, which is suppressed by a dominant-negative form of CHOP. Apoptosis triggered by ER stress may thus be involved in the development of NSAID-induced gastric lesions.
The induction of ER stress by NSAIDs is not specific to gastric mucosal cells. We found that indomethacin induced ER stress in murine podocytes [63]. It is known that NSAIDs cause nephrotoxicity, and ER stress may underlie the nephrotoxic potential of NSAIDs. Indeed, Lorz et al. reported that paracetamol (also known as acetaminophen) induces apoptosis of renal tubular cells and that it can be correlated with induction of ER stress as evidenced by up-regulation of CHOP and cleavage of procaspase-12 [64].
Immunosuppressants
Calcineurin inhibitors, cyclosporine A (CsA) and tacrolimus (FK506), improve allograft survival in organ transplantation. However, chronic allograft dysfunction caused by these drugs is a major hindrance to long-term graft survival. Nephrotoxicity of these agents may be one of major factors responsible for renal allograft dysfunction in the chronic phase [65]. Justo et al. reported that renal tubular apoptosis induced by CsA is associated with induction of CHOP [66]. We found that, in cultured renal tubular cells, CsA and FK506 caused upregulation of endogenous and exogenous indicators for ER stress. The induction of ER stress by these agents was shown to be reversible and observed similarly in other non-immune cells. Systemic administration with CsA into mice also causes rapid, significant induction of ER stress in the kidney [67]. Furthermore, Peyrou et al. reported that ER stress preconditioning was effective in decreasing the toxicity of CsA in renal tubular cell lines [68]. These results indicate a role of ER stress in the nephrotoxicity of calcineurin inhibitors.
Anti-cancer agents
A wide range of anti-cancer agents have the potential to induce ER stress. One of the most famous examples is cisplatin. Cisplatin is a chemotherapeutic agent used for the treatment of various solid tumors. Despite its therapeutic effectiveness, its nephrotoxic side effects limit its clinical use. Cisplatin has multiple intracellular effects including direct cytotoxicity through generation of ROS, activation of mitogen-activated protein kinases (MAP kinases), and stimulation of inflammation and fibrogenesis via generation of cytokines [69]. In renal tubular cells, cisplatin causes cleavage of procaspase-12, and blockade of caspase-12 activation significantly attenuates cisplatin-induced apoptosis [70]. Furthermore, ER stress preconditioning is effective in attenuating of the cisplatin cytotoxicity [68]. After administration with cisplatin in rats, activation of the XBP1 pathway and cleavage of procaspase-12 can be detected in the kidney [71]. These results suggest that ER stress-triggered activation of caspase-12 plays a pivotal role in cisplatin-induced nephrotoxicity. Currently, it is not fully understood how cisplatin induces ER stress, but ROS are considered as important mediators for cisplatin-induced tubular injury [69]. If so, the oxidative stress–ER stress pathway may be involved in the anti-tumor effect of cisplatin as well as its nephrotoxic side effects.
Proteasome inhibitors are currently used for the treatment of cancers [72]. Bortezomib is the first proteasome inhibitor approved by the US Food and Drug Administration for the treatment of multiple myeloma and lymphoma. The anti-cancer mechanisms of bortezomib include up-regulation of pro-apoptotic proteins, inhibition of NF-κB and its anti-apoptotic target genes, suppression of anti-apoptotic proteins, and downregulation of proteins involved in DNA repair [73]. In addition to these mechanisms, induction of ER stress may also play a role. Inhibition of the ubiquitin-proteasome pathway is a well-known trigger of ER stress via suppression of protein degradation and consequent accumulation of unfolded proteins in the ER. ER stress induced by proteasome inhibitors (bortezomib and next generation inhibitors, e.g., carfilzomib and marizomib) should be an additional important mechanism involved in their anti-cancer effects in general [73].
Imatinib mesylate (Gleevec) is a small-molecule inhibitor of the fusion protein Bcr-Abl, the causal molecule in chronic myelogenous leukemia. A previous report documented ten individuals who developed severe congestive heart failure during administration with imatinib [74]. Consistent with these observations in humans, imatinib-treated mice developed left ventricular contractile dysfunction. In the hearts of humans and mice treated with imatinib, accumulation of membrane whorls was observed in the ER, suggesting a toxic myopathy. With imatinib treatment, cardiomyocytes in culture exhibited ER stress as evidenced by activation of the PERK–eIF2α pathway and the IRE1–JNK pathway. Retroviral gene transfer of an imatinib-resistant mutant of c-Abl alleviated ER stress and rescued cardiomyocytes from imatinib-induced death [74], indicating a pathological role of ER stress in imatinib-induced cardiomyopathy.
Antibiotics
Aminoglycosides are powerful bactericidal antibiotics, but also major nephrotoxic antibiotics that cause renal tubular injury. The toxicity of aminoglycosides is related to their interference with the metabolism of anionic phospholipids, especially phosphoinositides. Jin et al. reported that a neomycin analogue geneticin caused cleavage of m-calpain and procaspase-12 in normal rat kidney cells, indicating involvement of ER stress [75]. Another report showed that, after administration with gentamicin in rats, activation of the XBP1 pathway and cleavage of procaspase-12 were induced in the kidney [71]. ER stress preconditioning was effective in attenuating the toxicity of gentamicin in LLC-PK1 cells [68]. These results suggest that ER stress is involved in aminoglycoside-induced renal injury.
Chlorhexidine is widely used as an antiseptic agent in medicine and dentistry. However, it may have a toxic effect in humans. A recent study showed that chlorhexidine elicited accumulation of proteins in the ER and expression of GRP78 in fibroblasts [76], leading to apoptotic cell death.
HIV protease inhibitors
Development and clinical use of HIV protease inhibitors have contributed greatly to the treatment of HIV. These agents are also used for the treatment of cancers. However, it is known that their therapeutic effect is accompanied by metabolic adverse effects including hyperlipidemia, insulin resistance, central fat accumulation and hepatic steatosis. Previous studies indicated that HIV protease inhibitors interfered with intracellular key processes regulating glucose and lipid metabolism in insulin-responsive tissues and that it was caused by induction of ER stress [77]. Several reports showed that HIV protease inhibitors including nelfinavir, ritonavir, lopinavir, and amprenavir elicited ER stress in normal and malignant cells [78–81]. It was evidenced by induction of ATF3 and ATF4, expression of GRP78 and CHOP, and activation of the PERK, ATF6 and IRE1 pathways. The induction of ER stress may be causative of their therapeutic effectiveness as well as their adverse effects on intact tissues.
Psychotropic drugs
Bipolar disorder is a common mood disorder characterized by recurrent episodes of mania and depression. Previous studies have suggested an involvement of ER stress responses in bipolar disorder. Genetic linkage studies and microarray analysis indicated that aberrant expression and/or function of XBP1 and GRP78 may be a risk factor for bipolar disorder [82, 83].
Valproate, a simple branched-chain fatty acid, has been used for the treatment of bipolar disorder, but the molecular mechanisms underlying its therapeutic effect are unclear. A previous review summarized the regulation of ER stress by valproate and discussed its possible implications in the therapeutic effects [84]. For example, members of the ER stress protein family (GRP78, GRP94, and calreticulin) are up-regulated by treatment with valproate. Immunohistochemistry showed that ER stress proteins were observed to increase in the frontal and parietal cortex, as well as in the hippocampus of rat brains following chronic treatment with valproate [84]. Another mood stabilizing drug lithium is also effective in the treatment of bipolar disorder. Like valproate, lithium induced expression of GRP78, GRP94, and calreticulin in cerebral cortical cells after its chronic treatment at therapeutically relevant concentrations [85]. These mood-stabilizing drugs could rescue impaired function of XBP1 and ER chaperones [83, 85]. Physiological levels of the UPR induced by valproate and lithium may correct abnormal ER stress conditions, leading to their therapeutic effectiveness in bipolar disorder.
Alcohol
Excessive alcohol consumption induces various pathogenic stress responses including the UPR [86]. ER stress contributes to alcoholic disorders in the liver, pancreas, heart, and brain. Potential mechanisms that trigger alcoholic ER stress are directly or indirectly related to alcohol metabolism which is associated with generation of toxic acetaldehyde and homocysteine, oxidative stress, perturbation of calcium or iron homeostasis, alteration in the S-adenosylmethionine to S-adenosylhomocysteine ratio, and abnormal epigenetic modifications [86].
Using subtractive hybridization to isolate ethanol-responsive genes, Miles et al. first reported that ethanol induced expression of GRP78 and GRP94 in a concentration-dependent manner [87]. Another report suggested that ethanol could increase GRP78 and GRP94 expression by altering ER calcium levels or glycoprotein trafficking [88].
Acute ethanol loading causes oxidative stress and activates JNK-mediated proapoptotic signaling in the liver. Using acute ethanol loading in perfused rats, Nishitani and Matsumoto reported that activation of JNK by ethanol was caused by ER stress [89]. Addition of antioxidant is reduced the activation of JNK, suggesting a link between oxidative stress and ER stress in ethanol-induced JNK activation and consequent apoptosis [89].
Acetaldehyde is an electrophilic substance endogenously produced as a first intermediate in oxidative ethanol metabolism. Ethanol impairs the mitochondrial transport of reduced glutathione (GSH), resulting in lower mitochondrial GSH (mGSH) levels. A previous report showed that acetaldehyde-treated HepG2 cells exhibited cytoplasmic lipid droplets and swollen mitochondria [90]. Acetaldehyde depleted the mGSH pool with spared cytosol GSH levels, which was caused by enhanced cholesterol deposition. The acetaldehyde-stimulated increase in mitochondrial cholesterol was preceded by induction of CHOP and sterol regulatory element-binding protein 1. ER stress-inducing agents mimicked the effect of acetaldehyde [90]. Acetaldehyde is a mediator for ER stress-triggered increases in lipid content in ethanol-exposed hepatocytes.
Physical stress
Acidic stress
Acidosis occurs under various pathophysiological situations and affects local and systemic cell function. Metabolic acidosis and respiratory acidosis are typical examples of systemic acidosis. An acidic milieu also develops locally as within ischemic lesions and tumors, and may determine cell survival and sensitivity to radiation and chemotherapy [91]. Aoyama et al. reported that transient acidosis induced ER stress and caspase-12-mediated cell death in mouse astrocytes. Acidosis increased expression of GRP78, phosphorylation of IRE1 and cleavage of procaspase-12, leading to activation of caspase-3 [92]. The authors showed that acidosis-induced ER stress may contribute to delayed cell death after ischemia.
Local acidosis occurs artificially during peritoneal dialysis. Peritoneal dialysis fluid (PDF) has several nonphysiological properties including high glucose and high lactate concentrations, hyperosmolarity and acidic pH (∼5.0). Previous reports have suggested that the parietal peritoneum from patients on peritoneal dialysis exhibit excessive development of the rough ER [93]. Another report also showed that rats with recurrent injections of PDF exhibited expansion of the ER in mesothelial cells [94]. These results suggest the possibility that PDF may induce ER overload in the mesothelium. Recently, we found that PDF, but not neutralized PDF, caused ER stress in the murine peritoneum. In vitro, acidic stress, but not metabolic and osmotic stress, induced ER stress in mesothelial cells and other cell types [95]. Currently, the molecular mechanisms underlying induction of ER stress by acidosis are unclear. A previous report showed that, in vascular smooth muscle cells, extracellular low pH induced Ca2+ release from the ER [96]. Because depletion of Ca2+ stores in the ER is a trigger for ER stress, it may be causative of the induction of ER stress by low pH. Ryanodine receptors might be responsible for the acidosis-triggered Ca2+ release from the ER [96].
Bacterial peritonitis is a frequent complication in patients on peritoneal dialysis. We reported that PDF suppressed expression of monocyte chemoattractant protein 1 (MCP-1) in mesothelial cells in vitro and in vivo and that it was ascribed to suppression of NF-κB [97]. ER stress has the potential to regulate NF-κB positively (early phase) or negatively (late phase) [50, 51]. We found that acidic stress suppressed activation of NF-κB and NF-κB-dependent MCP-1 induction in mesothelial cells [95]. This effect was mimicked by other ER stress inducers, and attenuation of ER stress reversed the suppressive effects of low pH on NF-κB. Furthermore, PDF and ER stress inducers suppressed expression of MCP-1 in the peritoneum in LPS-treated mice [95, 97]. These results indicate a role for the acidic stress–ER stress pathway in blunted activation of NF-κB, which may cause perturbations of monocyte recruitment by mesothelial cells and susceptibility to bacterial peritonitis in peritoneal dialysis patients.
Thermal stress
The ER chaperone GRP78 belongs to the 70 kDa heat shock protein family. However, GRP78 is not typically induced by thermal stress. However, some reports have demonstrated that heat shock may induce ER stress and upregulate GRP78 under particular circumstances. Xu et al. reported that exposure to mild hyperthermia (40 °C) induced ER stress responses including induction of GRP78, ERP72, DNAJC3, GADD34, and CHOP mRNAs, splicing of XBP1 mRNA and phosphorylation of eIF2α in mammalian cells [98]. Interestingly, this phenomenon was not observed when the cells were exposed to 43 °C. Using a rat model, Liu et al. also demonstrated that thermal stress activated ER stress, which may inhibit heat shock responses via translational block [99]. In heat-stressed rats, the UPR (eIF2α phosphorylation and XBP1 splicing) occurred mainly in the cerebral cortex, where the heat shock response was substantially inhibited. Inhibition of the heat shock response enhanced heat-induced ER stress and subsequent cell death [99]. These results imply crosstalk between thermal stress and ER stress.
UV light and X-ray
UVA
UV irradiation is a harmful electromagnetic wave. Because of the gradual thinning of the earth’s ozone layer, increased exposure to UV has become a serious problem for human health. UV is classified into three categories; UVA (wavelength, 315–400 nm), UVB (280–315 nm), and UVC (200–280 nm). UVA has the potential to cause damage to the epidermis and dermis; whereas, UVB affects only the epidermis. UVC hardly reaches human skin, because it is absorbed by the ozone layer. Because of this, UVA has been considered as a major factor responsible for photo-induced damage of the dermis. A recent report showed that UVA irradiation triggered conversion of p90ATF6 to an active transcription factor p50ATF6 in normal human dermal fibroblasts. UVA also induced IRE1-mediated splicing of XBP1 mRNA as well as PERK-mediated phosphorylation of eIF2α, leading to activation of ERSE-, UPRE-, and AARE-responsive targets [100]. Currently, it is not fully elucidated how UVA induces ER stress. However, UVA irradiation causes production of ROS [101]. Generation of ROS possibly mediates UVA-induced ER stress and contributes to inflammation and premature aging in the skin.
UVB and UVC
Like UVA, UVB may also have the potential to induce ER stress in keratinocytes. Using HaCaT cells, a recent report showed that environmental levels of UVB-induced ER stress. UVB induced nuclear translocation of ATF6 and induction of the IRE1–XBP1 pathway; whereas, PERK was not activated [102]. In contrast, another report showed that UVC activated the PERK–eIF2α pathway in COS-1, MCF-7, and HIT cells, leading to inhibition of protein synthesis [103].
X-rays
X-ray irradiation is used for cancer treatment, but its utility is often limited because of damage of normal tissues including the vascular endothelium. A recent study investigated the mechanism underlying X-ray-induced endothelial damage and found that 10 Gy induced ∼99.9 % loss of cell viability mainly due to apoptosis [104]. ER stress was evidenced by induction of GRP78, CHOP, and GADD34 as well as phosphorylation of eIF2α, and attenuation of ER stress by salubrinal blocked apoptosis by approximately 50 %. Like UVA, X-rays have the potential to trigger ROS production [105]. The oxidative stress–ER stress pathway may contribute to the induction of apoptosis by X-ray.
Hypoxic stress
Hypoxia triggers dramatic changes in cellular physiology by causing cell cycle arrest, a shift in metabolic balance and expression of proangiogenic factors. Although hypoxia-inducible factor 1 (HIF-1) controls expression of an array of hypoxia-regulated genes, HIF-1 activation alone cannot account for the full repertoire of changes that occur under hypoxia. Currently, it is well accepted that hypoxia causes ER stress, which contributes to gene expression, translational suppression and growth arrest that occur under hypoxic conditions [106]. The mechanisms responsible for the induction of ER stress by hypoxia are not fully elucidated. However, yeast uses a dedicated machinery of molecular chaperones and foldases to ensure proper folding of ER proteins. Foldases, such as protein disulfide isomerase and Ero1, catalyze the formation of disulfide linkages by transiently forming mixed disulfides with their client proteins and acting as an electron relay system for oxidative folding [107]. Molecular oxygen is the major electron acceptor at the end of this relay system, providing the driving force for protein folding in the ER. A similar mechanism of oxidative protein folding may exist in mammalian cells, and if so, accumulation of unfolded proteins in the ER is a natural consequence of hypoxia [106].
Hypoxia-induced ER stress is involved in the induction of cell death under hypoxic conditions. For example, reduced blood flow results in tissue hypoxia and hypoglycemia, which cause protein misfolding and ER stress. Reperfusion of the ischemic tissues triggers oxidative stress, and resultant ROS and NO further enhance ER stress. ROS and NO may also modify oxidizable residues (cysteines) in ER-associated Ca2+ channels, leading to depletion of Ca2+ and consequent protein misfolding. These molecular events contribute to ischemia–reperfusion injury. Previous reports have shown that, in brain ischemia, activation of the PERK–eIF2α pathway induced CHOP that played a causal role in neuronal cell death. NO, a known mediator of brain injury during stroke, may be involved in this process [108].
In tumors, however, activation of the PERK–eIF2α pathway may contribute to tumor cell survival. It is known that development of chronic and fluctuating hypoxic regions results in malignant progression and tolerance to therapy in tumors. Hypoxia induces phosphorylation of eIF2α via PERK, and inactivation of either PERK or eIF2α impairs tumor cell survival under hypoxia. PERK- or eIF2α-inactivated tumors show slow growth in nude mice and higher levels of apoptosis in hypoxic areas. The UPR pathway mediated by IRE1 and its downstream target XBP1 may also be required for hypoxia tolerance in tumors in vitro and in vivo [109].
Malnutrition
In mammals, plasma concentrations of nutrients decline under nutritional or pathological conditions, which may cause ER stress. The most typical examples are glucose and amino acid starvation. Deficiency in some micronutrients such as vitamins may also induce the UPR.
Glucose
Cells respond to glucose limitation by decreasing global protein synthesis and increasing synthesis of selective proteins that affect cell survival. Induction of the UPR is one of the mechanisms involved in this process. Glucose limitation is known to induce the UPR by altering protein glycosylation in the ER [1]. Glucose deprivation causes activation of the PERK–eIF2α pathway, which may contribute to global translational suppression and induction of some genes including CHOP [110].
Amino acids
It is known that amino acids are involved in the control of some genes including CHOP. Bruhat et al. examined molecular mechanisms involved in the regulation of CHOP upon amino acid limitation. The authors showed that a cis element, AARE, was essential for amino acid regulation of the CHOP gene promoter. They also showed that the CHOP-AARE element was related to C/EBP and ATF/CRE binding sites and bound ATF2, which was essential for the transcriptional activation of CHOP induced by amino acid starvation [111]. On the other hand, Jian et al. reported that expression of ATF3 was required for the induction of CHOP by amino acid starvation, and it was mediated by the PERK–eIF2α–ATF4 pathway [112].
Vitamins
Thiamine (vitamin B1) deficiency causes region-selective neuronal loss in the brain. Wang et al. reported that thiamine deficiency up-regulated expression of GRP78 and CHOP, phosphorylation of eIF2α and cleavage of procaspase-12 in the cerebellum and the thalamus of mice. Treatment of cultured cerebellar granule neurons with an inhibitor of thiamine transport caused ER stress and consequent apoptosis. It was associated with generation of ROS [113].
Secretory proteins undergo oxidative folding (formation of disulfide bonds) in the ER before secretion. Oxidative folding depends on flavoproteins in eukaryotes. Manthey et al. reported that when HepG2 cells were cultured in riboflavin (vitamin B2)-deficient medium, secretion of apolipoprotein B-100 significantly decreased. Nuclear translocation of ATF6 and consequent expression of GRP78 were induced in riboflavin-deficient cells. Phosphorylation of eIF2α and down-stream induction of CHOP also increased in riboflavin-deficient cells [114].
Ascorbate (vitamin C) is produced in the liver or kidney of most animals, whereas some species including humans and guinea pigs are deficient in gulonolactone oxidase, the enzyme required for catalizing the last step of ascorbate synthesis. Insufficient intake of vitamin C in these species, therefore, causes scurvy, a disease characterized by extreme weakness and various skin and gum abnormalities. It has been suggested that ascorbate participates in oxidative protein folding in the ER. Using a ascorbate-deficient model in guinea pigs, Margittai et al. reported that persistent ascorbate deficiency led to the UPR (expression of GRP78 and GRP94) and apoptosis in the liver. The authors suggested that ER stress may participate in the pathology of scurvy [115].
Tocotrienols are naturally occurring forms of vitamin E and have the potential to induce apoptosis in a variety of cancer cells. Park et al. reported that γ-tocotrienol increased the level of ER stress markers in mammary cancer cells. They also showed that γ-tocotrienol activated JNK and p38 MAP kinase, events downstream of IRE1 activation, and up-regulated death receptor 5 (DR5) and CHOP. Silencing either JNK or p38 MAP kinase reduced the induction of DR5 and CHOP and partially attenuated γ-tocotrienol-induced apoptosis. The authors concluded that up-regulation of DR5 and CHOP by γ-tocotrienol was dependent on JNK and p38 MAP kinase activation mediated by ER stress [116]. The link between apoptosis and ER stress in γ-tocotrienol-treated cancer cells has also been documented by other investigators [117].
ER stress and environmental factor-triggered pathologies: an oxidative stress–ER stress axis
As I have described, a wide range of environmental factors have the potential to induce ER stress. Although underlying mechanisms are different from factor to factor, a number of environmental pollutants trigger both ER stress and oxidative stress. Those include toxic metals, cigarette smoke, paraquat, silica nanoparticle, TBTO and nitro-PAH. LPS, anticancer agents, alcohol, UV, hypoxia and vitamin deficiency also induce ER stress in parallel with oxidative stress, as described in this article. Currently, the relationship between oxidative stress and ER stress has not been fully elucidated.
Previous reports have shown that ROS is generated in renal tubular cells in response to cadmium [19, 118–120]. When the cells were treated with antioxidants, induction of apoptosis by cadmium was attenuated [19, 119]. Haynes et al. reported that prolonged activation of the UPR results in oxidative stress and consequently cellular death, indicating that oxidative stress may be an event downstream of ER stress [121]. However, accumulating evidence suggests that activation of the UPR occurs upon exposure to oxidative stress, and it is an adaptive mechanism to preserve cell function and survival, as reviewed by Malhotra and Kaufman [122]. We found that cadmium-induced ER stress was attenuated by antioxidants in renal tubular cells. Exposure of the cells to ROS donors caused ER stress. In contrast, suppression of ER stress by overexpression of GRP78 or 150 kDa oxygen-regulated protein did not attenuate cadmium-triggered oxidative stress, suggesting that ER stress is the event downstream of oxidative stress [19].
The phenomenon described above may be not specific to the particular cell type and the particular stimulus. We found that exposure of bronchial epithelial cells to cigarette smoke caused ER stress and apoptosis, and attenuation of ER stress by overexpression of ER chaperones significantly suppressed the apoptotic event. Exposure of the cells to cigarette smoke caused the generation of ROS, and treatment with antioxidants also inhibited cigarette smoke-induced apoptosis [22]. As in cadmium-exposed renal tubular cells, antioxidants blunted the induction of CHOP in cigarette smoke-exposed bronchial epithelial cells, suggesting ER stress mediated apoptosis is downstream of oxidative stress [22].
As described, ER stress is the crucial event downstream of oxidative stress in cadmium-induced apoptosis. However, there is limited information available regarding which ROS plays the major roles in the induction of ER stress and apoptosis. Using renal tubular epithelial cells, we showed that exposure to O2 ·−, H2O2 or ONOO− induced apoptosis, whereas ER stress (indicated by expression of GRP78, GRP94, and CHOP) was caused only by O2 ·− and ONOO−. Transfection with manganese superoxide dismutase, the scavenger of O2 ·−, significantly attenuated cadmium-induced ER stress and apoptosis, whereas pharmacological inhibition of ONOO− was ineffective. Interestingly, overexpression of catalase attenuated cadmium-induced apoptosis partially without affecting the level of ER stress [19]. These results suggest that O2 ·− is selectively involved in cadmium-triggered, ER stress-mediated apoptosis of renal tubular cells.
Using bronchial epithelial cells, we further investigated the roles of individual ROS in the induction of CHOP by cigarette smoke [24]. Exposure of bronchial epithelial cells to O2 ·−, ONOO− or H2O2 induced expression of CHOP, whereas, NO alone did not. Induction of CHOP by cigarette smoke was attenuated by scavengers for O2 ·−, ONOO−, or NO, whereas scavenging H2O2 did not affect the induction of CHOP. Cigarette smoke, O2 ·−- and ONOO−-induced phosphorylation of PERK and eIF2α, and dominant-negative inhibition of the PERK–eIF2α pathway suppressed cigarette smoke-triggered induction of CHOP and apoptosis [24]. These results suggest that O2 ·− and ONOO− are selectively involved in cigarette smoke-triggered induction of CHOP and that the PERK–eIF2α pathway plays a crucial role in the induction of CHOP and apoptosis downstream of particular ROS.
How does oxidative stress induce ER stress? Previous reports showed that oxidative stress causes inhibition of Ca2+-ATPase [123, 124]. ROS may induce depletion of calcium stores in the ER via inhibition of Ca2+-ATPase [125] and thereby cause ER stress. Another possibility is that ROS might cause ER stress through generation and accumulation of oxidatively modified, abnormal proteins. Unfolded proteins might also accumulate in the ER through ROS-induced functional perturbation of foldases and/or chaperones in the ER [126].
In general, ROS has the potential to induce ER stress. The fact that a wide range of environmental pollutants trigger generation of ROS [127] indicates that the oxidative stress–ER stress axis is an important mechanism underlying induction of ER stress by environmental factors.
Conclusions
ER stress and consequent UPR are triggered by various environmental factors including pollutants, infectious pathogens, drugs, alcohol, physical stress and malnutrition. The ER stress that is induced contributes to the self-defense of local tissues or induction of tissue injury in ER stress-related disorders including heavy metal intoxication, cigarette smoke-related disorders, infectious diseases, ischemic injury, adverse effects of drugs and alcohol-induced tissue damage. Although the mechanisms underlying the induction of ER stress are different from factor to factor, a number of environmental factors trigger the generation of ROS. The oxidative stress–ER stress axis may be a pivotal mechanism in the induction of ER stress by environmental factors.
Abbreviations
- ER:
-
Endoplasmic reticulum
- UPR:
-
Unfolded protein response
- ERAD:
-
ER-associated degradation
- PERK:
-
RNA-dependent protein kinase-like ER kinase
- ATF6:
-
Activating transcription factor 6
- IRE1:
-
Inositol-requiring enzyme 1
- eIF2α:
-
Eukaryotic translation initiation factor 2α
- XBP1:
-
X-box binding protein 1
- ERSE:
-
ER stress response element
- UPRE:
-
UPR element
- GRP78:
-
78 kDa glucose-regulated protein
- CHOP:
-
CCAAT/enhancer-binding protein-homologous protein
- AARE:
-
Amino acid response element
- TRAF2:
-
Tumor necrosis factor receptor-associated factor 2
- ASK1:
-
Apoptosis signal-regulating kinase 1
- JNK:
-
c-Jun N-terminal kinase
- PAH:
-
Polycyclic aromatic hydrocarbon
- HIV:
-
Human immunodeficiency virus
- UV:
-
Ultraviolet
- ROS:
-
Reactive oxygen species
- mTORC1:
-
Mammalian target of rapamycin complex 1
- COPD:
-
Chronic obstructive pulmonary disease
- RNS:
-
Reactive nitrogen species
- TBTO:
-
Bis(tri-n-butyltin)oxide
- 1-NP:
-
1-nitropyrene
- LPS:
-
Lipopolysaccharide
- NF-κB:
-
Nuclear factor-κB
- SubAB:
-
Subtilase cytotoxin
- NSAID:
-
Non-steroidal anti-inflammatory drug
- CsA:
-
Cyclosporine A
- FK506:
-
Tacrolimus
- MAP kinase:
-
Mitogen-activated protein kinase
- GSH:
-
Glutathione
- PDF:
-
Peritoneal dialysis fluid
- MCP-1:
-
Monocyte chemoattractant protein 1
- HIF-1:
-
Hypoxia-inducible factor 1
- DR5:
-
Death receptor 5
References
Lee AS (2000) The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci 26:504–510
Kaufman RJ (2002) Orchestrating the unfolded protein response in health and disease. J Clin Invest 110:1389–1398
Wu J, Kaufman RJ (2006) From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ 13:374–384
Kim R, Emi M, Tanabe K, Murakami S (2006) Role of the unfolded protein response in cell death. Apoptosis 11:5–13
Jeong SH, Habeebu SS, Klaassen CD (2000) Cadmium decreases gap junctional intercellular communication in mouse liver. Toxicol Sci 57:156–166
Thevenod F (2003) Nephrotoxicity and the proximal tubule. Insights from cadmium. Nephron Physiol 93:87–93
Yokouchi M, Hiramatsu N, Hayakawa K, Kasai A, Takano Y, Yao J, Kitamura M (2007) Atypical, bidirectional regulation of cadmium-induced apoptosis via distinct signaling of unfolded protein response. Cell Death Differ 14:1467–1474
Liu F, Inageda K, Nishitai G, Matsuoka M (2006) Cadmium induces the expression of Grp78, an endoplasmic reticulum molecular chaperone, in LLC-PK1 renal epithelial cells. Environ Heal Perspect 114:859–864
Hiramatsu N, Kasai A, Du S, Takeda M, Hayakawa K, Okamura M, Yao J, Kitamura M (2007) Rapid, transient induction of ER stress in the liver and kidney after acute exposure to heavy metal: Evidence from transgenic sensor mice. FEBS Lett 581:2055–2059
Qian Y, Falahatpisheh MH, Zheng Y, Ramos KS, Tiffany-Castiglioni E (2001) Induction of 78 kD glucose-regulated protein (GRP78) expression and redox-regulated transcription factor activity by lead and mercury in C6 rat glioma cells. Neurotox Res 3:581–589
Stacchiotti A, Morandini F, Bettoni F, Schena I, Lavazza A, Grigolato PG, Apostoli P, Rezzani R, Aleo MF (2009) Stress proteins and oxidative damage in a renal derived cell line exposed to inorganic mercury and lead. Toxicology 264:215–224
Lu TH, Su CC, Chen YW, Yang CY, Wu CC, Hung DZ, Chen CH, Cheng PW, Liu SH, Huang CF (2011) Arsenic induces pancreatic β-cell apoptosis via the oxidative stress-regulated mitochondria-dependent and endoplasmic reticulum stress-triggered signaling pathways. Toxicol Lett 201:15–26
Zhang R, Piao MJ, Kim KC, Kim AD, Choi JY, Choi J, Hyun JW (2012) Endoplasmic reticulum stress signaling is involved in silver nanoparticles-induced apoptosis. Int J Biochem Cell Biol 44:224–232
Wang Z, Wang H, Xu ZM, Ji YL, Chen YH, Zhang ZH, Zhang C, Meng XH, Zhao M, Xu DX (2012) Cadmium-induced teratogenicity: association with ROS-mediated endoplasmic reticulum stress in placenta. Toxicol Appl Pharmacol 259:236–247
Zhang Y, Sun LG, Ye LP, Wang B, Li Y (2008) Lead-induced stress response in endoplasmic reticulum of astrocytes in CNS. Toxicol Mech Methods 18:751–757
Shinkai Y, Yamamoto C, Kaji T (2010) Lead induces the expression of endoplasmic reticulum chaperones GRP78 and GRP94 in vascular endothelial cells via the JNK-AP-1 pathway. Toxicol Sci 114:378–386
Prozialeck WC, Lamar PC (2005) Effects of glutathione depletion on the cytotoxic actions of cadmium in LLC-PK1 cells. Toxicol. Appl Pharm 134:285–295
Thevenod F, Friedmann JM, Katsen AD, Hauser IA (2005) Up-regulation of multidrug resistance P-glycoprotein via NF-κB activation protects kidney proximal tubule cells from cadmium- and reactive oxygen species-induced apoptosis. J Biol Chem 275:1887–1896
Yokouchi M, Hiramatsu N, Hayakawa K, Okamura M, Du S, Kasai A, Takano Y, Shitamura A, Shimada T, Yao J, Kitamura M (2008) Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response. J Biol Chem 283:4252–4260
Tiffany-Castiglioni E, Qian Y (2012) ER chaperone-metal interactions: links to protein folding disorders. Neurotoxicology 33:545–557
Malhotra D, Thimmulappa R, Vij N, Navas-Acien A, Sussan T, Merali S, Zhang L, Kelsen SG, Myers A, Wise R, Tuder R, Biswal S (2009) Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: the role of Nrf2-regulated proteasomal activity. Am J Respir Crit Care Med 180:1196–1207
Tagawa Y, Hiramatsu N, Kasai A, Hayakawa K, Okamura M, Yao J, Kitamura M (2008) Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP). Free Radic Biol Med 45:50–59
Church DF, Pryor WA (1985) Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Heal Perspect 64:111–126
Tagawa Y, Hiramatsu N, Kato H, Sakoh T, Nakajima S, Hayakawa K, Saito Y, Johno H, Takahashi S, Gu L, Yao J, Kitamura M (2011) Induction of CCAAT/enhancer-binding protein-homologous protein by cigarette smoke through the superoxide anion-triggered PERK–eIF2α pathway. Toxicology 287:105–112
Baker RR (2006) Environ Toxicol. Environ Toxicol 6:621–622
Haberzettl P, Vladykovskaya E, Srivastava S, Bhatnagar A (2008) Role of endoplasmic reticulum stress in acrolein-induced endothelial activation. Toxicol Appl Pharmacol 234:14–24
Kitaguchi Y, Taraseviciene-Stewart L, Hanaoka M, Natarajan R, Kraskauskas D, Voelkel NF (2012) Acrolein induces endoplasmic reticulum stress and causes airspace enlargement. PLoS One 7:e38038
Smith CJ, Perfetti TA, Morton MJ, Rodgman A, Garg R, Selassie CD, Hansch C (2002) The relative toxicity of substituted phenols reported in cigarette mainstream smoke. Toxicol Sci 69:265–278
Dinis-Oliveira RJ, Remião F, Carmo H, Duarte JA, Navarro AS, Bastos ML, Carvalho F (2006) Paraquat exposure as an etiological factor of Parkinson's disease. Neurotoxicology 27:1110–1122
Chinta SJ, Rane A, Poksay KS, Bredesen DE, Andersen JK, Rao RV (2008) Coupling endoplasmic reticulum stress to the cell death program in dopaminergic cells: effect of paraquat. Neuromol Med 10:333–342
Yang W, Tiffany-Castiglioni E, Koh HC, Son IH (2002) Paraquat activates the IRE1/ASK1/JNK cascade associated with apoptosis in human neuroblastoma SH-SY5Y cells. Toxicol Lett 191:203–210
Suntres ZE (2009) Role of antioxidants in paraquat toxicity. Toxicology 180:65–77
Napierska D, Thomassen LC, Lison D, Martens JA, Hoet PH (2010) The nanosilica hazard: another variable entity. Part Fibre Toxicol 7:39
Christen V, Fent K (2012) Silica nanoparticles and silver-doped silica nanoparticles induce endoplasmatic reticulum stress response and alter cytochrome P4501A activity. Chemosphere 87:423–434
Luo FC, Zhou J, Lv T, Qi L, Wang SD, Nakamura H, Yodoi J, Bai J (2012) Induction of endoplasmic reticulum stress and the modulation of thioredoxin-1 in formaldehyde-induced neurotoxicity. Neurotoxicology 33:290–298
Katika MR, Hendriksen PJ, van Loveren H, Peijnenburg A (2011) Exposure of Jurkat cells to bis(tri-n-butyltin)oxide (TBTO) induces transcriptomics changes indicative for ER- and oxidative stress, T cell activation and apoptosis. Toxicol Appl Pharmacol 254:311–322
van Kol SW, Hendriksen PJ, van Loveren H, Peijnenburg A (2012) Transcriptomics analysis of primary mouse thymocytes exposed to bis(tri-n-butyltin)dioxide (TBTO). Toxicology 296:37–47
Andersson H, Piras E, Demma J, Hellman B, Brittebo E (2009) Low levels of the air pollutant 1-nitropyrene induce DNA damage, increased levels of reactive oxygen species and endoplasmic reticulum stress in human endothelial cells. Toxicology 262:57–64
Nakagomi T, Kitada O, Yoshikawa K, Ozawa K, Ogawa S, Matsuyama T (2004) The 150-kilodalton oxygen-regulated protein ameliorates lipopolysaccharide-induced acute lung injury in mice. Am J Pathol 165:1279–1288
Endo M, Oyadomari S, Suga M, Mori M, Gotoh T (2005) The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice. J Biochem 138:501–507
Hiramatsu N, Kasai A, Hayakawa K, Yao J, Kitamura M (2006) Real-time detection and continuous monitoring of ER stress in vitro and in vivo by ES-TRAP: evidence for systemic, transient ER stress during endotoxemia. Nucleic Acids Res 34:e93
Nakayama Y, Endo M, Tsukano H, Mori M, Oike Y, Gotoh T (2010) Molecular mechanisms of the LPS-induced non-apoptotic ER stress-CHOP pathway. J Biochem 147:471–483
Kim HJ, Kim SR, Park JK, Kim DI, Jeong JS, Lee YC (2012) PI3Kγ activation is required for LPS-induced reactive oxygen species generation in respiratory epithelial cells. Inflamm Res 61:1265–1272
Pahl HL, Baeuerle PA (1996) Activation of NF-κB by ER stress requires both Ca2+ and reactive oxygen intermediates as messengers. FEBS Lett 392:129–136
Zhang K, Kaufman R (2008) From endoplasmic-reticulum stress to the inflammatory response. Nature 454:455–462
Hayakawa K, Hiramatsu N, Okamura M, Yamazaki H, Nakajima S, Yao J, Paton AW, Paton JC, Kitamura M (2009) Acquisition of anergy to proinflammatory cytokines in non-immune cells through endoplasmic reticulum stress response: a mechanism for subsidence of inflammation. J Immunol 182:1182–1191
Hayakawa K, Nakajima S, Hiramatsu N, Okamura M, Huang T, Saito Y, Tagawa Y, Tamai M, Takahashi S, Yao J, Kitamura M (2010) ER stress depresses NF-κB activation in mesangial cells through preferential induction of C/EBPβ. J Am Soc Nephrol 21:73–81
Nakajima S, Hiramatsu N, Hayakawa K, Saito Y, Kato H, Huang T, Yao J, Paton AW, Paton JC, Kitamura M (2011) Selective abrogation of BiP/GRP78 blunts activation of NF-κB through the ATF6 branch of the UPR: involvement of C/EBPβ and mTOR-dependent dephosphorylation of Akt. Mol Cell Biol 31:1710–1718
Harama D, Koyama K, Mukai M, Shimokawa N, Miyata M, Nakamura Y, Ohnuma Y, Ogawa H, Matsuoka S, Paton AW, Paton JC, Kitamura M, Nakao A (2009) A sub-cytotoxic dose of subtilase cytotoxin prevents LPS-induced inflammatory responses, depending on its capacity to induce the unfolded protein response. J Immunol 183:1368
Kitamura M (2009) Biphasic, bidirectional regulation of NF-κB by endoplasmic reticulum stress. Antioxid Redox Signal 11:2353–2364
Kitamura M (2011) Control of NF-κB and inflammation by the unfolded protein response. Int Rev Immunol 30:4–15
Tesh VL (2012) Activation of cell stress response pathways by Shiga toxins. Cell Microbiol 14:1–9
Lee S-Y, Lee M-S, Cherla RP, Tesh VL (2008) Shiga toxin 1 induces apoptosis through the ER stress response in human monocytic cells. Cell Microbiol 10:770–780
Paton AW, Srimanote P, Talbot UM, Wang H, Paton JC (2004) A new family of potent AB5 cytotoxins produced by Shiga toxigenic Escherichia coli. J Exp Med 200:35–46
Paton AW, Paton JC (2005) Multiplex PCR for direct detection of Shiga toxigenic Escherichia coli producing the novel subtilase cytotoxin. J Clin Microbiol 43:2944–2947
Paton AW, Beddoe T, Thorpe CM, Whisstock JC, Wilce MC, Rossjohn J, Talbot UM, Paton JC (2006) AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 443:548–552
Wolfson JJ, May KL, Thorpe CM, Jandhyala DM, Paton JC, Paton AW (2008) Subtilase cytotoxin activates PERK, IRE1 and ATF6 endoplasmic reticulum stress-signalling pathways. Cell Microbiol 10:1775–1786
Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R, Huang T, Nakajima S, Yao J, Paton AW, Paton JC, Kitamura M (2009) Activation of the Akt–NF-κB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol 183:1480–1487
He B (2006) Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ 13:393–403
Yoshida H (2007) ER stress and diseases. FEBS J 274:630–658
Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C (2003) Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J 22:5435–5445
Tsutsumi S, Gotoh T, Tomisato W, Mima S, Hoshino T, Hwang HJ, Takenaka H, Tsuchiya T, Mori M, Mizushima T (2004) Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ 11:1009–1016
Okamura M, Takano Y, Hiramatsu N, Hayakawa K, Yao J, Paton AW, Paton JC Kitamura M (2008) Suppression of cytokine responses by indomethacin in podocytes: a mechanism through induction of unfolded protein response. Am J Physiol Renal Physiol 295:F1495–F1503
Lorz C, Justo P, Sanz A, Subirá D, Egido J, Ortiz A (2004) Paracetamol-induced renal tubular injury: a role for ER stress. J Am Soc Nephrol 15:380–389
Williams D, Haragsim L (2006) Calcineurin nephrotoxicity. Adv Chronic Kidney Dis 13:47–55
Justo P, Lorz C, Sanz A, Egido J, Ortiz A (2003) Intracellular mechanisms of cyclosporin A-induced tubular cell apoptosis. J Am Soc Nephrol 14:3072–3080
Du S, Hiramatsu N, Hayakawa K, Kasai A, Okamura M, Huang T, Yao J, Takeda M, Araki I, Sawada N, Paton AW, Paton JC, Kitamura M (2009) Suppression of NF-κB by cyclosporine A and tacrolimus (FK506) via induction of the C/EBP family: implication for unfolded protein response. J Immunol 182:7201–7211
Peyrou M, Cribb AE (2007) Effect of endoplasmic reticulum stress preconditioning on cytotoxicity of clinically relevant nephrotoxins in renal cell lines. Toxicol in Vitro 21:878–886
Yao X, Panichpisal K, Kurtzman N, Nugent K (2007) Cisplatin nephrotoxicity: a review. Am J Med Sci 334:115–124
Liu H, Bowes R (2005) Endoplasmic reticulum stress-associated caspase-12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J Am Soc Nephrol 16:1985–1992
Peyrou M, Hanna PE, Cribb AE (2007) Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicol Sci 99:346–353
Adams J, Palombella VJ, Elliott PJ (2000) Proteasome inhibition: a new strategy in cancer treatment. Invest New Drugs 18:109–1021
Mujtaba T, Dou QP (2011) Advances in the understanding of mechanisms and therapeutic use of bortezomib. Discov Med 12:471–480
Kerkelä R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, Rosenzweig A, Salomon RN, Van Etten RA, Alroy J, Durand JB, Force T (2006) Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med 12:908–916
Jin QH, Zhao B, Zhang XJ (2004) Cytochrome c release and endoplasmic reticulum stress are involved in caspase-dependent apoptosis induced by G418. Cell Mol Life Sci 61:1816–1825
Faria G, Cardoso CR, Larson RE, Silva JS, Rossi MA (2009) Chlorhexidine-induced apoptosis or necrosis in L929 fibroblasts: a role for endoplasmic reticulum stress. Toxicol Appl Pharmacol 234:256–265
Parker RA, Flint OP, Mulvey R, Elosua C, Wang F, Fenderson W, Wang S, Yang WP, Noor MA (2005) Endoplasmic reticulum stress links dyslipidemia to inhibition of proteasome activity and glucose transport by HIV protease inhibitors. Mol Pharmacol 67:1909–1919
Pyrko P, Kardosh A, Wang W, Xiong W, Schönthal AH, Chen TC (2007) HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res 67:10920–10928
Kraus M, Malenke E, Gogel J, Müller H, Rückrich T, Overkleeft H, Ovaa H, Koscielniak E, Hartmann JT, Driessen C (2008) Ritonavir induces endoplasmic reticulum stress and sensitizes sarcoma cells toward bortezomib-induced apoptosis. Mol Cancer Ther 7:1940–1948
Chen L, Jarujaron S, Wu X, Sun L, Zha W, Liang G, Wang X, Gurley EC, Studer EJ, Hylemon PB, Pandak WM Jr, Zhang L, Wang G, Li X, Dent P, Zhou H (2009) HIV protease inhibitor lopinavir-induced TNF-α and IL-6 expression is coupled to the unfolded protein response and ERK signaling pathways in macrophages. Biochem Pharmacol 78:70–77
Wu X, Sun L, Zha W, Studer E, Gurley E, Chen L, Wang X, Hylemon PB, Pandak WM Jr, Sanyal AJ, Zhang L, Wang G, Chen J, Wang JY, Zhou H (2010) HIV protease inhibitors induce endoplasmic reticulum stress and disrupt barrier integrity in intestinal epithelial cells. Gastroenterology 138:197–209
Kakiuchi C, Ishiwata M, Nanko S, Kunugi H, Minabe Y, Nakamura K, Mori N, Fujii K, Umekage T, Tochigi M, Kohda K, Sasaki T, Yamada K, Yoshikawa T, Kato T (2005) Functional polymorphisms of HSPA5: possible association with bipolar disorder. Biochem Biophys Res Commun 336:1136–1143
Kakiuchi C, Iwamoto K, Ishiwata M, Bundo M, Kasahara T, Kusumi I, Tsujita T, Okazaki Y, Nanko S, Kunugi H, Sasaki T, Kato T (2003) Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat Genet 35:171–175
Bown CD, Wang JF, Chen B, Young LT (2002) Regulation of ER stress proteins by valproate: therapeutic implications. Bipolar Disord 4:145–151
Shao L, Sun X, Xu L, Young LT, Wang JF (2006) Mood stabilizing drug lithium increases expression of endoplasmic reticulum stress proteins in primary cultured rat cerebral cortical cells. Life Sci 78:1317–1323
Ji C (2013) Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem Res Int (in press)
Miles MF, Wilke N, Elliot M, Tanner W, Shah S (1994) Ethanol-responsive genes in neural cells include the 78-kilodalton glucose-regulated protein (GRP78) and 94-kilodalton glucose-regulated protein (GRP94) molecular chaperones. Mol Pharmacol 46:873–879
Hsieh KP, Wilke N, Harris A, Miles MF (1996) Interaction of ethanol with inducers of glucose-regulated stress proteins. Ethanol potentiates inducers of grp78 transcription. J Biol Chem 271:2709–2716
Nishitani Y, Matsumoto H (2006) Ethanol rapidly causes activation of JNK associated with ER stress under inhibition of ADH. FEBS Lett 580:9–14
Lluis JM, Colell A, García-Ruiz C, Kaplowitz N, Fernández-Checa J (2003) Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology 124:708–724
Reichert M, Steinbach JP, Supra P, Weller M (2002) Modulation of growth and radiochemosensitivity of human malignant glioma cells by acidosis. Cancer 95:1113–1119
Aoyama K, Burns DM, Suh SW, Garnier P, Matsumori Y, Shiina H, Swanson RA (2012) Acidosis causes endoplasmic reticulum stress and caspase-12-mediated astrocyte death. J Cereb Blood Flow Metab 25:358–370
Dobbie JW (1989) Morphology of the peritoneum in CAPD. Blood Purif 7:74–85
Slater ND, Cope GH, Raftery AT (1991) Mesothelial hyperplasia in response to peritoneal dialysis fluid: a morphometric study in the rat. Nephron 58:466–471
Johno H, Ogata R, Nakajima S, Hiramatsu N, Kobayashi T, Hara H, Kitamura M (2012) Acidic stress–ER stress axis for blunted activation of NF-κB in mesothelial cells exposed to peritoneal dialysis fluid. Nephrol Dial Transplant 27:4053–4060
Rohra DK, Saito SY, Ohizumi Y (2003) Mechanism of acidic pH-induced contraction in spontaneously hypertensive rat aorta: role of Ca2+ release from the sarcoplasmic reticulum. Acta Physiol Scand 179:273–280
Ogata R, Hiramatsu N, Hayakawa K, Nakajima S, Yao J, Kobayashi T, Kitamura M (2012) Impairment of MCP-1 expression in mesothelial cells exposed to PDF by osmotic stress and acidic stress. Perit Dial Int 31:80–89
Xu X, Gupta S, Hu W, McGrath BC, Cavener DR (2011) Hyperthermia induces the ER stress pathway. PLoS One 6:e23740
Liu Y, Sakamoto H, Adachi M, Zhao S, Ukai W, Hashimoto E, Hareyama M, Ishida T, Imai K, Shinomura Y (2012) Heat stress activates ER stress signals which suppress the heat shock response, an effect occurring preferentially in the cortex in rats. Mol Biol Rep 39:3987–3993
Komori R, Taniguchi M, Ichikawa Y, Uemura A, Oku M, Wakabayashi S, Higuchi K, Yoshida H (2012) Ultraviolet a induces endoplasmic reticulum stress response in human dermal fibroblasts. Cell Struct Funct 37:49–53
Tyrrell RM (2004) Solar ultraviolet A radiation: an oxidizing skin carcinogen that activates heme oxygenase-1. Antioxid Redox Signal 6:835–840
Mera K, Kawahara K, Tada K, Kawai K, Hashiguchi T, Maruyama I, Kanekura T (2012) ER signaling is activated to protect human HaCaT keratinocytes from ER stress induced by environmental doses of UVB. Biochem Biophys Res Commun 397:350–354
Wu S, Hu Y, Wang JL, Chatterjee M, Shi Y, Kaufman RJ (2002) Ultraviolet light inhibits translation through activation of the unfolded protein response kinase PERK in the lumen of the endoplasmic reticulum. J Biol Chem 277:18077–18083
Panganiban RA, Mungunsukh O, Day RM (2013) X irradiation induces ER stress, apoptosis, and senescence in pulmonary artery endothelial cells. Int J Radiat Biol (in press)
Datta R, Hallahan DE, Kharbanda SM, Rubin E, Sherman ML, Huberman E, Weichselbaum RR, Kufe DW (1992) Involvement of reactive oxygen intermediates in the induction of c-Jun gene transcription by ionizing radiation. Biochemistry 31:8300–8306
Feldman DE, Chauhan V, Koong AC (2005) The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol Cancer Res 3:597–605
Tu BP, Weissman JS (2004) Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 164:341–346
Xu C, Bailly-Maitre B, Reed JC (2005) Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115:2656–2664
Koumenis C (2006) ER stress, hypoxia tolerance and tumor progression. Curr Mol Med 6:55–69
Fernandez J, Bode B, Koromilas A, Diehl JA, Krukovets I, Snider MD, Hatzoglou M (2002) Translation mediated by the internal ribosome entry site of the cat-1 mRNA is regulated by glucose availability in a PERK kinase-dependent manner. J Biol Chem 277:11780–11787
Bruhat A, Jousse C, Carraro V, Reimold AM, Ferrara M, Fafournoux P (2000) Amino acids control mammalian gene transcription: activating transcription factor 2 is essential for the amino acid responsiveness of the CHOP promoter. Mol Cell Biol 20:7192–7204
Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC (2004) Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol 24:1365–1377
Wang X, Wang B, Fan Z, Shi X, Ke ZJ, Luo J (2007) Thiamine deficiency induces endoplasmic reticulum stress in neurons. Neuroscience 144:1045–1056
Manthey KC, Chew YC, Zempleni J (2012) Riboflavin deficiency impairs oxidative folding and secretion of apolipoprotein B-100 in HepG2 cells, triggering stress response systems. J Nutr 135:978–982
Margittai E, Bánhegyi G, Kiss A, Nagy G, Mandl J, Schaff Z, Csala M (2005) Scurvy leads to endoplasmic reticulum stress and apoptosis in the liver of Guinea pigs. J Nutr 135:2530–2534
Park SK, Sanders BG, Kline K (2010) Tocotrienols induce apoptosis in breast cancer cell lines via an endoplasmic reticulum stress-dependent increase in extrinsic death receptor signaling. Breast Cancer Res Treat 124:361–375
Wali VB, Bachawal SV, Sylvester PW (2009) Endoplasmic reticulum stress mediates gamma-tocotrienol-induced apoptosis in mammary tumor cells. Apoptosis 14:1366–1377
Prozialeck WC, Lamar PC (1995) Effects of glutathione depletion on the cytotoxic actions of cadmium in LLC-PK1 cells. Toxicol Appl Pharmacol 134:285–295
Thevenod F, Friedmann JM, Katsen AD, Hauser IA (2000) Up-regulation of multidrug resistance P-glycoprotein via NF-κB activation protects kidney proximal tubule cells from cadmium- and reactive oxygen species-induced apoptosis. J Biol Chem 275:1887–1896
Gennari A, Cortese E, Boveri M, Casado J, Prieto P (2003) Sensitive endpoints for evaluating cadmium-induced acute toxicity in LLC-PK1 cells. Toxicology 183:211–220
Haynes CM, Titus EA, Cooper AA (2004) Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell 15:767–776
Malhotra JD, Kaufman RJ (2007) Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 9:2277–22793
Kaplan P, Babusikova E, Lehotsky J, Dobrota D (2003) Free radical-induced protein modification and inhibition of Ca2+-ATPase of cardiac sarcoplasmic reticulum. Mol Cell Biochem 248:41–47
Moreau VH, Castilho RF, Ferreira ST, Carvalho-Alves PC (1998) Oxidative damage to sarcoplasmic reticulum Ca2+-ATPase AT submicromolar iron concentrations: evidence for metal-catalyzed oxidation. Free Radic Biol Med 25:554–560
Viner RI, Huhmer AF, Bigelow DJ, Schoneich C (1996) The oxidative inactivation of sarcoplasmic reticulum Ca2+-ATPase by peroxynitrite. Free Radic Res 24:243–259
Brunet S, Thibault L, Lepage G, Seidman EG, Dube N, Levy E (2000) Modulation of endoplasmic reticulum-bound cholesterol regulatory enzymes by iron/ascorbate-mediated lipid peroxidation. Free Radic Biol Med 28:46–54
Ahmad S (1995) Oxidative stress from environmental pollutants. Arch Insect Biochem Physiol 29:135–157
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on “The unfolded protein response in immune diseases” - Guest Editors: Richard Blumberg and Arthur Kaser
Rights and permissions
About this article
Cite this article
Kitamura, M. The unfolded protein response triggered by environmental factors. Semin Immunopathol 35, 259–275 (2013). https://doi.org/10.1007/s00281-013-0371-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-013-0371-y