
Abstract
The question if enteroviruses could cause beta-cell damage and type 1 diabetes has become more and more relevant when recent studies have provided new evidence supporting this scenario. One important observation is the recent discovery of IFIH1 as a risk gene for type 1 diabetes. This gene is an innate immune system receptor for enteroviruses offering one possible mechanism for the diabetogenic effect of enteroviruses. This is further emphasized by the observations suggesting that the innate immune system is activated in the pancreatic islets of type 1 diabetic patients and that the innate immune system is important for the defense against the virus and for the regulation of adaptive immune system. Important progress has also been gained in studies analyzing pancreas tissue for possible presence of enteroviruses. Several studies have found enteroviruses in the pancreatic islets of type 1 diabetic patients using various methods. The virus seems to be located in the islets while exocrine pancreas is mostly uninfected. One recent study found the virus in the intestinal mucosa in the majority of diabetic patients. Enteroviruses can also infect cultured human pancreatic islets causing either rapid cell destruction or a persistent-like noncytolytic infection. Combined with all previous, epidemiological findings indicating the risk effect of enteroviruses in cross-sectional and prospective studies, these observations fit to a scenario where certain diabetogenic enterovirus variants establish persistent infection in gut mucosa and in the pancreatic islets. This in turn could lead to a local inflammation and the breakdown of tolerance in genetically susceptible individuals. This is also supported by mouse experiments showing that enteroviruses can establish prolonged infection in the pancreas and intestine, and some virus strains cause beta-cell damage and diabetes. In conclusion, recent studies have strengthened the hypothesis that enteroviruses play a role in the pathogenesis of type 1 diabetes. These findings open also new opportunities to explore the underlying mechanism and get closer to causal relationship.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The enterovirus (EV) genus is part of the large picornavirus family. They are small non-enveloped RNA viruses which are the most common viruses causing human diseases. Usually, they lead to mild or asymptomatic infections, but occasionally, the virus spreads to the myocardium or central nervous system leading to myocarditis, meningitis, or paralysis. Infections are common already in very young infants, and newborns are at risk to get systemic multiorgan infections, which can be fatal.
Type 1 diabetes (T1D) is caused by a selective destruction of insulin producing pancreatic beta-cells. Many risk factors are known, such as the HLA genes, but the triggers, which induce the autoimmune process leading to beta-cell death, are still largely unknown. However, evidence supporting the role of enteroviruses can be seen as one of the strongest. These are for example: 1) the risk effect of EVs has been documented in most cross-sectional and prospective studies; 2) EV proteins and viral RNA have been detected in the pancreas and intestinal mucosa of T1D patients; 3) EVs have strong tropism to human pancreatic islets but not to the exocrine pancreas; 4) certain EV receptors (CAR) are strongly expressed in human pancreatic islets but not in exocrine pancreas; 5) antiviral defense mechanisms are activated in the pancreatic islets of T1D patients; 6) EVs infect and damage beta-cells in cultured human pancreatic islets; 7) the innate immune system receptor for EVs (IFIH1) is a risk gene for T1D; 8) EVs activate an inflammatory process and cause diabetes in animal models; 9) EVs establish persistent infection in pancreatic islets and gut mucosa in mouse models; and 10) the seasonality of autoantibody appearance and of the onset of T1D follows a similar seasonal pattern as EVs.
Epidemiological studies
A considerable body of evidence has suggested that EV infections are involved in the pathogenesis of T1D, and this association has long been implied from epidemiological evidence. Original studies were done by Gamble and Taylor [23, 24] who found that the seasonal pattern of T1D overlapped that of EVs showing a peak in autumn. In addition, they found that enterovirus antibodies were more frequent in T1D patients than in control subjects. Since then, enterovirus antibodies have been measured from T1D patients and control subjects in several studies with varying results [36, 53]. Overall, these studies suggest that T1D patients may indeed have an increased frequency of enterovirus antibodies, but the difference to control subjects is not very clear, and results vary according to the method used for antibody measurements, the population which was examined and the selection of control subjects. It should also be noted that we may not expect to see clear differences in antibody frequency between case and control subjects if the diabetogenic virus is frequent in the background population and causes beta-cell damage only in a small proportion of infected individuals.
In addition to serological studies, EVs have been searched for using molecular methods. Detection of the virus directly from blood by sensitive RT-PCR assay has proved to be a valuable tool in diabetes research. At least nine cross-sectional studies have been published where EV genome has been found in blood of T1D patients (Table 1) [1, 7–9, 21, 39, 46, 47, 77]. This method has made it possible also to type the virus by sequencing its genome. However, the sequences obtained from blood of T1D patients, so far, cover only the conserved region of the EV genome (5′NCR), which does not allow detailed typing of the virus strain. Altogether, these studies have indicated that about 30% of T1D patients are positive for EV RNA in blood or serum compared to less than 5% in control subjects. This suggests that T1D patients have more enterovirus infections or the immune response is weaker in T1D patients than control subjects, which could lead to prolonged infections in T1D patients. It is not known if these infections represent an acute or persistent infection. In acute infections, EV is present in serum only for a short period of time, usually no more than a few days, which decreases the possibility of virus detection. On the other hand, in persistent infection, the amount of virus in serum is probably very small, which may limit the possibilities to detect such infection even with highly sensitive PCR techniques. Furthermore, used cDNA amplification methods are usually designed only for positive strand RNA and therefore these methods do not detect effectively negative strand RNA. It has been shown that in persistent infections negative strand RNA is sometimes incorrectly packed into the viral capsids [41]. Thus, it is possible that these studies underestimate the frequency of virus in T1D patients.
Prospective studies offer several advantages compared to cross-sectional studies. Such studies are based on a follow-up of initially non-diabetic subjects until some of them develop T1D. An important advantage is that one can analyze time-relationship between virus infection and the initiation of the autoimmune process reflected by the appearance of islet autoantibodies in serum. The first prospective studies were carried out in Finland, where EVs were found more frequently in children who later progressed to T1D than in control children and infections clustered to the time when islet antibodies appeared [32, 35]. These findings have been repeated in further prospective studies in the Finnish population using both antibody measurements and EV RNA detection from serum [43, 44, 56, 57, 60, 61]. However, prospective studies in the US and Germany could not find this risk effect [22, 27]. It is not known if this could be due to methodological differences (e.g., much longer sample intervals in the US and German series) or true differences between populations. Altogether, the prospective studies done so far are based on relatively small series of children, and larger studies in different populations are obviously needed. This is a rapidly expanding research field and large international studies, such as the TEDDY study, are currently in progress. In conclusion, the data from epidemiological studies suggests that the presence of EV in blood is associated with increased risk of T1D. It seems that the presence of EV in stools is not an as clear risk marker as EV in blood. This may indicate that systemic infection or viral persistence in certain internal organs is an important factor in EV induced beta-cell damage.
In addition to childhood infections, EV infections during pregnancy may increase the risk of T1D in the offspring [10–12, 35]. However, the risk effect of maternal infections during pregnancy has not been found in all studies [74]. Altogether, it seems that the role of such infections needs further confirmation and if it exists it may be relatively small or it exists in some subpopulations.
If EVs cause T1D, one can ask if they could also contribute to the increasing incidence of T1D. For example, the frequency of EV infections, particularly that caused by diabetogenic strains, could have increased during the past decades paralleling the increase in T1D incidence. This question was evaluated in a series of studies carried out in the EPIVIR project. The EPIVIR project included several European countries with varying incidences of T1D. Possible change in EV infection frequency over the last decades was analyzed by measuring EV antibodies in stored serum samples taken from pregnant women during the years 1983–2001 in Finland and Sweden. Interestingly, EV antibodies decreased significantly during this time period suggesting that EV infections may actually have became less frequent [71, 73]. The antibody assays that were used in the EPIVIR project detected antibodies against a wide range of different EV serotypes as a group, suggesting that the overall incidence of EV infections has decreased. However, this does not exclude the possibility that a certain specific EV type could follow an opposite pattern and an increasing trend. The finding of EPIVIR project led to the generation of a new hypothesis, the polio hypothesis. The name of this hypothesis refers to a similar epidemiological change in paralytic polio about 100 years ago. Polio is caused by three EV serotypes, polioviruses 1–3, which destroy motoneurons in the spinal cord leading to paralysis in about 1% of infected individuals. The incidence of this paralytic complication started to increase rapidly at the end of the 19th century (polio epidemics started) at the same time when the circulation of poliovirus decreased. This resembles the current epidemiological findings in T1D which increases at the same time when EV are becoming less frequent. The increase in paralytic polio was due to a shift in the child's first infections from neonatal period to an older age when maternal antibodies had already disappeared from the child's circulation and were not protecting the child. According to the polio hypothesis the same phenomenon happens now in T1D when the protection of infants against diabetogenic EV variants has decreased due to the decrease in maternal antibody frequency and the delay in the infections to an older age. The same phenomenon may also contribute to the marked difference in the incidence of T1D between countries. The findings of the EPIVIR project suggest that the frequency of EV infections is lower in countries with high incidence of T1D (such as Finland) compared to that of countries with low incidence of T1D [71, 72]. Similar difference was seen in the history of paralytic polio, which was more frequent in the USA than in Egypt, while an opposite trend was seen in the frequency of poliovirus infections. Currently, it is not known if the polio hypothesis is true for T1D, but it clearly forms an interesting basis for further studies on infection ecology and T1D.
Another hypothesis that could link EVs to the increasing incidence of T1D is the hygiene hypothesis. In contrast to the polio hypothesis, which deals with immune protection against EVs, hygiene hypothesis postulates that EVs may have a beneficial effect on immune regulation and down-regulate autoimmune responses by so-called bystander suppression mechanism. This idea was supported by experiments in NOD mice, as EVs can prevent spontaneously developing diabetes in these mice when they are infected early, before insulitis has started [18, 70]. This protection is mediated at least partly by the induction of regulatory T cells, but it is not specific for EVs, as many other viruses, including diabetogenic encephalomyocarditis virus EMCV-D strain, can also prevent diabetes in NOD mice [31]. EVs are also associated with low risk of IgE-mediated allergic sensitization [64] supporting the idea that they may have such immunoregulatory effects also in man.
Viral determinants of diabetogenicity
EMCV infection in mice has widely been used to study the mechanisms of picornavirus-induced T1D. Diabetogenic EMCV variant (EMCV-D) causes diabetes by damaging insulin producing beta-cells while another variant (EMCV-B) does not. Diabetogenic strain may induce lower interferon response than non-diabetogenic strain leading to more severe infection in the pancreatic islets [3]. These two virus variants differ in several amino acids, but a single point mutation in the major capsid protein VP1 region seems to be the major determinant of diabetogenicity [38]. The location of this amino acid in the receptor binding region suggests that it may modulate the binding affinity of the virus to its cellular receptor. In analogy with EMCV-D certain EV strains have also been cited as diabetogenic. These strains have usually been isolated from T1D patients, and some of them can cause diabetes in mice. One of the most widely used such strains is the E2 strain of coxsackievirus B4 [80]. However, the molecular determinants of diabetogenicity of EVs have not been identified. Experience from other EV diseases, particularly poliomyelitis, indicates that EV virulence can be controlled by several genomic regions including the 5′NCR and the capsid region. The identification of such determinants would be important for the understanding of the mechanisms of EV-induced beta-cell damage in man. They could be related to the binding of the virus to its cellular receptors, viral interactions with intracellular proteins during viral replication cycle or the effect of the virus on the immune system. Critical bottlenecks hindering the progress in this field include the limited number of potentially diabetogenic wild-type EV strains isolated from diabetic and prediabetic subjects as well as the high mutation and recombination rate of EVs that cause a lot of natural variability in these viruses and leads to the generation of viral quasispecies during infection. In addition, it is not known how well different mouse models predict the diabetogenicity of different EV strains in man. Recently, a systematic collection of picornavirus strains from prediabetic and diabetic children has been started in Finland as a part of international collaboration (PicoBank) aiming at identification of diabetogenic determinants by analyzing a large number of potentially diabetogenic and non-diabetogenic EV strains and comparing their properties to those of previously reported diabetogenic strains.
Role of host factors
It is known from other EV diseases that host factors regulate the outcome of EV infection. For example, the male gender increases the risk of severe EV illness both in man (e.g. paralysis, meningitis, myocarditis) and it increases also the risk of picornavirus-induced diabetes in mice [40, 76]. Young age is another risk factor for severe disease as newborns are highly susceptible for coxsackievirus B infections. Dietary factors also play a role and e.g., the deficiency of selenium is associated with severe course of EV infection (coxsackievirus-induced myocarditis) [2]. It has been shown that breast-feeding protects against EV infections [59]. EVs may also interact with some cow's milk components, such as cow's milk insulin, leading to stronger diabetogenic effect in infants who have received cow's milk formula than in breast-fed infants [45].
Animal experiments have demonstrated that the individual genetic setup of the host has a clear influence on the development of beta-cell damage and diabetes after viral infection. This is clearly indicated by the fact that only certain mouse strains develop diabetes after picornavirus infection [42, 55, 81]. This susceptibility is partly associated with MHC genes but also other genes are involved. Genome-wide association studies have identified at least 40 regions in human genome that are associated with human T1D, and many of them are known to be relevant for antiviral responses. For example, the HLA locus, which includes the major susceptibility alleles for T1D, regulates the course of many virus infections including dengue, hepatitis C, HIV and hantaan virus infection. HLA risk genes for T1D modulate immune response against EVs and strong antibody response against EVs is associated with HLA-DR3 and HLA-DR4 risk antigens [58]. The significance of this association is not known, but it is possible that strong antiviral response leads to more severe tissue pathology in infected islets. Recently, the IFIH1/MDA5 gene was found to mediate susceptibility for T1D [66, 67, 75]. Rare IFIH1 gene variants that modulate the structure of the IFIH1 are strongly associated with protection against T1D and also exert an influence on IFIH1–mRNA expression in peripheral blood mononuclear cells (PBMCs) [48]. Other mutations in the IFIH1 region have also shown association with T1D. The IFIH1 gene codes for an important innate immune system sensor for EVs and some other picornaviruses such as EMCV, detecting viral double-stranded RNA (dsRNA), which is produced in the cytoplasm during virus replication. IFIH1-deficient mice are also highly susceptible to infections caused by these viruses [34]. Thus, this molecule offers a plausible mechanistic explanation for the previously observed link between the innate immune system, EV infection and susceptibility to T1D. As the innate immune response is important for the clearance of infection and for the regulation of adaptive immunity, it is quite obvious that this finding opens new opportunities to study the mechanisms mediating the risk effect of EVs. It is possible that IFIH1-mediated activation of innate immune system by EVs determines if the virus can invade pancreatic islets and/or if it can cause strong enough inflammation to break the immune tolerance.
Pancreas studies
Recently, a causal relationship has received important support from studies showing EV RNA and proteins in the pancreas of T1D patients (Table 2) [52, 79]. In a recent study, EV VP1 protein was found by immunohistochemistry (IHC) in the pancreas of 61% of recent-onset T1D patients but in only 6% of relevant controls [52]. This result was confirmed using an antibody against dsRNA-activated protein kinase R (PKR). Similar findings have come from recent case reports and from the new Network for Pancreatic Organ Donors with Diabetes (nPOD) project (Table 2) [13, 26, 50, 69, 80]. Also, there is now direct evidence of an enterovirus infection in pancreatic beta-cells in fulminant diabetes [65, 68]. Although there has been evidence of the role of virus infections in the etiology of fulminant T1D, these recent papers are the first studies where enteroviruses were detected directly in the pancreas. Importantly, all these studies have found the virus almost exclusively in the pancreatic islets while exocrine pancreas has been virus negative, a pattern which parallels islet-specific expression of one major EV receptor, CAR [50]. Support for the islet specificity of EVs has also been obtained from newborn babies who died of systemic EV infection. Also in these cases EV were found almost exclusively in pancreatic islets, while exocrine tissue has been mostly EV negative [19, 20, 37, 79]. In addition, EV has been found in the intestinal mucosa in the majority (75%) of T1D patients but in only 10% of control subjects [49]. Examples of EV-positive pancreatic islets and gut biopsies are shown in Fig. 1. Collectively, these studies suggest that T1D may be quite frequently associated with EV infection in the pancreatic islets and intestinal mucosa. In theory, such a high frequency of the virus in T1D patients would fit well with viral persistence in these tissues and suggests that it may spread from the intestinal mucosa to pancreas via common lymphatic and vascular networks of these closely located organs. Evidence for virus has been found using different methods (in situ hybridization, immunohistochemistry, and electron microscopy), and EVs have also been isolated from the pancreas of T1D patients [13, 80], which argues against methodological artifacts. Furthermore, one of these viruses was able to induce diabetes in mice. These findings are also in line with in vitro studies showing that human islets are highly permissive for EVs and that these viruses have a wide range of effects on islet cells ranging from persistence to rapid lytic infection depending on the type and dose of the virus [54, 78]. However, it is not known if some of these infection patterns are typical for diabetogenic EV strains.

EV positivity in pancreas (a,b) and duodenum (c,d), by immunohistochemistry (a,c) and in situ hybridization (b,d). Positivity to EVs is shown by a brown color in IHC stained tissues (a,c) and by a purple color in ISH (b,d). Arrows pointed EV positive cells. Magnification 400×
Positivity for EVs has been found to be located in beta-cells in most studies where double staining was done for EV protein and specific islet-cell proteins in pancreatic tissue [13, 52]. An example of such beta-cell-specific EV staining is shown in Fig. 2. The beta-cell-specific staining for EV has also been seen in beta-cells infected in cell cultures, although in some studies also non-beta-cells have been EV positive [6, 16]. The beta-cell specificity of EVs may vary between EV serotypes and possibly between virus strains, as different enterovirus types use different cellular receptors for entry.

Immunofluorescence double staining of EV proteins (green) and different pancreatic islet cell proteins (red) including insulin (a), glucagon (b) and somatostatin (c). Co-localization of EV and beta-cells is shown in yellow (a. Magnification 200×. Photo by Teppo Haapaniemi
One of the major obstacles in studying the diabetic pancreas has been and still is the shortage of tissues, both from diabetics and representative controls. One reason for this is the difficulty to take pancreatic biopsies, due to organ location and the risk of complications. Furthermore, there is practically no tissue available from autoantibody positive subjects, excluding some rare exceptions [25, 50]. Lately, this obstacle has been overcome due to screening of brain-dead organ donors for diabetes-associated autoantibodies. Although this effort, these tissues are still rare and require a large and expensive organization. Examples of these are the PanFin network in Finland, the nPOD in the USA, and Belgian Organ Donor Consortium.
Methodological limitations of the detection of enteroviruses in tissue samples
The most widely used detection method in tissue analysis is the IHC staining using the Dako EV antibody against EV VP1 protein (clone 5D8/1). Although this antibody has its weaknesses, it still seems to be the only commercial antibody, which properly detects EVs by IHC from formalin-fixed paraffin-embedded (FFPE) tissues. One limitation is that in certain conditions, EV antibodies might cross-react with some islet-cell antigens (IA-2) and HSP60 [28, 29]. It also might give background staining in smooth muscle cells [52]. However, by careful optimization, this antibody can be used reliably. However, it would still be important to have another method or antibody to confirm the result. Available methods include in situ hybridization and RT-PCR, both of which can be applied for FFPE samples. Electron microscopy (EM) and virus isolation need frozen samples and well-preserved tissue. Altogether, reliable detection of EVs from tissue samples is not a trivial issue, and positive findings should be confirmed using various methods. Every method however has its limitations, which are presented in Table 3.
Role of viral persistence
Persistence of a virus usually leads to an inflammatory process, which in many occasions is indistinguishable from an autoimmune process. A good example of such process is a chronic hepatitis C virus infection, which causes a strong inflammation process in the liver and induces an autoimmune response against liver antigens. Animal experiments have shown definitively that picornaviruses can cause persistent infections leading to severe tissue pathology. The classical model is the Theiler’s murine encephalomyelitis virus (TMEV) infection in mice. The DA strain of TMEV causes a persistent central nervous system infection in certain susceptible mouse strains, which is associated with restricted viral gene expression and yielding an inflammatory demyelizating response [15]. Class I genes within the MHC locus (H-2D region) play a major role in determining whether strains of mice develop chronic demyelization and TMEV persistence [4]. In addition to TMEV, EVs can persist for several months in the intestine, heart, and central nervous system in mice leading to tissue pathology such as myocarditis. For example, coxsackievirus B3 persists in a terminally deleted defective form in the myocardium. The pathogenetic mechanism seems to include both direct effects of viral replication as well as induction of inflammation in the heart [4]. EVs can also establish persistent infections in cell lines. Human studies have suggested that persistent EV infection may play a role in post polio syndrome, dilated cardiomyopathy and chronic fatigue syndrome as well as in the pathogenesis of T1D. This creates a challenge in the detection of such infection in T1D patients, since the replication of such virus may be at an extremely low level. The balance between positive and negative strand viral RNA may also be shifted from strong predominance of positive strand RNA (typical for acute infection) to equal amounts of positive and negative strand RNA (persistent infection). This, in turn, increases the formation of dsRNA complexes. EV variants, which can cause persistence have been shown to carry deletions in the 5′NCR of their genome [5, 17, 41]. Altogether, these characters can decrease the sensitivity of conventional assays to detect EV in T1D.
How enteroviruses break the tolerance
EV can cause an autoimmune process by various mechanisms. Studies in experimental animals have evaluated these mechanisms in detail and demonstrated that there is a clear biological basis for such phenomenon. However, it is still open which of these mechanisms could be relevant for human type 1 diabetes. One of the most likely possibilities is that the inflammation induced by EVs leads to an autoimmune response by so-called bystander activation mechanism [33]. In this scenario, the virus creates a proinflammatory milieu in infected pancreatic islets, which activates dendritic cells to present beta-cell autoantigens delivered from virus-damaged cells. EV infection in the pancreatic islets also increases the expression of HLA class I molecules by inducing type I interferons [51]. This may increase the activity of CD8+ cytotoxic T cells. Other innate immune system responses may also be important and especially the role of NK cells has been suggested [13, 14]. Immunological cross-reactivity between EV and pancreatic islets has also been described, which may contribute the beta-cell damaging process [29, 30]. Recent studies have implicated that antibody mediated enhancement of infection, a mechanism which mediates severe illness in some other virus diseases, such as dengue, may play a role in EV-induced T1D [62]. This phenomenon is based on antibodies that target the virus but which do not neutralize it. Such antibodies can worsen the course of disease probably by increasing the replication of EVs in monocytes/macrophages and by causing a shift from Th1 type immune response to Th2 type response.
The pathogenesis of fulminant T1D fits with an acute infection leading to a fast and complete destruction of beta-cells, since the disease progresses very fast. In fulminant diabetes also the amount of alpha-cells is reduced [63] and one can speculate that this might also be a consequence of the virus infection. Would the differences between autoimmune and fulminant T1D be due to different serotype of EVs causing the disease or to different immune response and genetic setup of the host. This is also an interesting but still open issue.
Conclusions
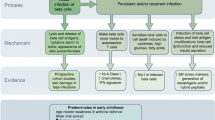
In conclusion, the present knowledge fits well to a model where certain diabetogenic EVs infect individuals at a young age leading to viral persistence in gut mucosa and in pancreatic islets. Virus may persist as a low-level, noncytolytic infection, causing local and long-lasting inflammation in the islets. This, in turn, could lead to the activation of dendritic cells in infected tissues and in the draining lymph nodes, which, together with virus-induced islet-cell damage, leads to the breakdown of tolerance in individuals who are genetically susceptible for T1D (Fig. 3). Studies of pancreatic tissues suggest that such EV strains may have strong tropism to islet cells especially to beta-cells. However, it is not known if these EVs represent certain genotypes or serotypes. From the therapeutic point of view, the information about the serotypes and genotypes of diabetogenic EVs would be crucial as it would directly facilitate the development of effective vaccines against these viruses. More than 100 different EV serotypes exist, and it is not known which of them are infecting the pancreatic islets or generating the risk effect observed in epidemiological studies. Large prospective studies in different populations as well as studies on the pancreatic tissue will be extremely valuable for making progress in these fundamental questions. Such studies, for example DIPP study (http://research.utu.fi/dipp/index.php?mid=2&language=en), TEDDY study (http://teddy.epi.usf.edu/) and nPOD study (http://www.jdrfnpod.org/) are currently in progress.

Hypothetical model of the pathogenesis of enterovirus-induced beta-cell damage. The figure illustrates how enterovirus infection could lead to an autoimmune process and beta-cell damage in pancreatic islets. This model is based on the combination of epidemiological evidence and virus analyses of tissue samples. Key component is prolonged or persistent infection in gut mucosa and pancreatic islets
References
Andreoletti L, Hober D, Hober-Vandenberghe C, Belaich S, Vantyghem MC, Lefebvre J, Wattre P (1997) Detection of coxsackie B virus RNA sequences in whole blood samples from adult patients at the onset of type I diabetes mellitus. J Med Virol 52:121–127
Beck MA, Levander OA, Handy J (2003) Selenium deficiency and viral infection. J Nutr 133:1463S–1467S
Cerutis DR, Bruner RH, Thomas DC, Giron DJ (1989) Tropism and histopathology of the D, B, K, and MM variants of encephalomyocarditis virus. J Med Virol 29:63–69
Chapman NM, Kim KS (2008) Persistent coxsackievirus infection: enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr Top Microbiol Immunol 323:275–292
Chapman NM, Kim KS, Drescher KM, Oka K, Tracy S (2008) 5′ terminal deletions in the genome of a coxsackievirus B2 strain occurred naturally in human heart. Virology 375:480–491
Chehadeh W, Kerr-Conte J, Pattou F, Alm G, Lefebvre J, Wattre P, Hober D (2000) Persistent infection of human pancreatic islets by coxsackievirus B is associated with alpha interferon synthesis in beta cells. J Virol 74:10153–10164
Chehadeh W, Weill J, Vantyghem MC, Alm G, Lefebvre J, Wattre P, Hober D (2000) Increased level of interferon-alpha in blood of patients with insulin-dependent diabetes mellitus: relationship with coxsackievirus B infection. J Infect Dis 181:1929–1939
Clements GB, Galbraith DN, Taylor KW (1995) Coxsackie B virus infection and onset of childhood diabetes. Lancet 346:221–223
Craig ME, Howard NJ, Silink M, Rawlinson WD (2003) Reduced frequency of HLA DRB1*03-DQB1*02 in children with type 1 diabetes associated with enterovirus RNA. J Infect Dis 187:1562–1570
Dahlquist GG, Ivarsson S, Lindberg B, Forsgren M (1995) Maternal enteroviral infection during pregnancy as a risk factor for childhood IDDM. A population-based case-control study. Diabetes 44:408–413
Dahlquist GG, Boman JE, Juto P (1999) Enteroviral RNA and IgM antibodies in early pregnancy and risk for childhood-onset IDDM in offspring. Diabetes Care 22:364–365
Dahlquist GG, Forsberg J, Hagenfeldt L, Boman J, Juto P (2004) Increased prevalence of enteroviral RNA in blood spots from newborn children who later developed type 1 diabetes: a population-based case-control study. Diabetes Care 27:285–286
Dotta F, Censini S, van Halteren AG, Marselli L, Masini M, Dionisi S, Mosca F, Boggi U, Muda AO, Prato SD, Elliott JF, Covacci A, Rappuoli R, Roep BO, Marchetti P (2007) Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci USA 104:5115–5120
Dotta F, Fondelli C, Falorni A (2008) Can NK cells be a therapeutic target in human type 1 diabetes? Eur J Immunol 38:2961–2963
Drescher KM, Sosnowska D (2008) Being a mouse in a man’s world: what TMEV has taught us about human disease. Front Biosci 13:3775–3785
Elshebani A, Olsson A, Westman J, Tuvemo T, Korsgren O, Frisk G (2007) Effects on isolated human pancreatic islet cells after infection with strains of enterovirus isolated at clinical presentation of type 1 diabetes. Virus Res 124:193–203
Feuer R, Ruller CM, An N, Tabor-Godwin JM, Rhoades RE, Maciejewski S, Pagarigan RR, Cornell CT, Crocker SJ, Kiosses WB, Pham-Mitchell N, Campbell IL, Whitton JL (2009) Viral persistence and chronic immunopathology in the adult central nervous system following Coxsackievirus infection during the neonatal period. J Virol 83:9356–9369
Filippi CM, Estes EA, Oldham JE, von Herrath MG (2009) Immunoregulatory mechanisms triggered by viral infections protect from type 1 diabetes in mice. J Clin Invest 119:1515–1523
Foulis AK, Farquharson MA, Cameron SO, McGill M, Schonke H, Kandolf R (1990) A search for the presence of the enteroviral capsid protein VP1 in pancreases of patients with type 1 (insulin-dependent) diabetes and pancreases and hearts of infants who died of coxsackieviral myocarditis. Diabetologia 33:290–298
Foulis AK, McGill M, Farquharson MA, Hilton DA (1997) A search for evidence of viral infection in pancreases of newly diagnosed patients with IDDM. Diabetologia 40:53–61
Foy CA, Quirke P, Lewis FA, Futers TS, Bodansky HJ (1995) Detection of common viruses using the polymerase chain reaction to assess levels of viral presence in type 1 (insulin-dependent) diabetic patients. Diabet Med 12:1002–1008
Fuchtenbusch M, Irnstetter A, Jager G, Ziegler AG (2001) No evidence for an association of coxsackie virus infections during pregnancy and early childhood with development of islet autoantibodies in offspring of mothers or fathers with type 1 diabetes. J Autoimmun 17:333–340
Gamble DR, Kinsley ML, FitzGerald MG, Bolton R, Taylor KW (1969) Viral antibodies in diabetes mellitus. Br Med J 3:627–630
Gamble DR, Taylor KW (1969) Seasonal incidence of diabetes mellitus. Br Med J 3:631–633
Gianani R, Putnam A, Still T, Yu L, Miao D, Gill RG, Beilke J, Supon P, Valentine A, Iveson A, Dunn S, Eisenbarth GS, Hutton J, Gottlieb P, Wiseman A (2006) Initial results of screening of non-diabetic organ donors for expression of islet autoantibodies. J Clin Endocrinol Metab 91:1855–1861
Gladisch R, Hofmann W, Waldherr R (1976) Myocarditis and insulitis following coxsackie virus infection. Z Kardiol 65:837–849
Graves PM, Rotbart HA, Nix WA, Pallansch MA, Erlich HA, Norris JM, Hoffman M, Eisenbarth GS, Rewers M (2003) Prospective study of enteroviral infections and development of beta-cell autoimmunity. Diabetes autoimmunity study in the young (DAISY). Diabetes Res Clin Pract 59:51–61
Harkonen T, Puolakkainen M, Sarvas M, Airaksinen U, Hovi T, Roivainen M (2000) Picornavirus proteins share antigenic determinants with heat shock proteins 60/65. J Med Virol 62:383–391
Harkonen T, Lankinen H, Davydova B, Hovi T, Roivainen M (2002) Enterovirus infection can induce immune responses that cross-react with beta-cell autoantigen tyrosine phosphatase IA-2/IAR. J Med Virol 66:340–350
Harkonen T, Paananen A, Lankinen H, Hovi T, Vaarala O, Roivainen M (2003) Enterovirus infection may induce humoral immune response reacting with islet cell autoantigens in humans. J Med Virol 69:426–440
Hermitte L, Vialettes B, Naquet P, Atlan C, Payan MJ, Vague P (1990) Paradoxical lessening of autoimmune processes in non-obese diabetic mice after infection with the diabetogenic variant of encephalomyocarditis virus. Eur J Immunol 20:1297–1303
Hiltunen M, Hyoty H, Knip M, Ilonen J, Reijonen H, Vahasalo P, Roivainen M, Lonnrot M, Leinikki P, Hovi T, Akerblom HK (1997) Islet cell antibody seroconversion in children is temporally associated with enterovirus infections. Childhood Diabetes in Finland (DiMe) Study Group. J Infect Dis 175:554–560
Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N (1998) Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med 4:781–785
Huhn MH, McCartney SA, Lind K, Svedin E, Colonna M, Flodstrom-Tullberg M Melanoma differentiation-associated protein-5 (MDA-5) limits early viral replication but is not essential for the induction of type 1 interferons after Coxsackievirus infection. Virology
Hyoty H, Hiltunen M, Knip M, Laakkonen M, Vahasalo P, Karjalainen J, Koskela P, Roivainen M, Leinikki P, Hovi T et al (1995) A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes 44:652–657
Hyoty H (2004) Environmental causes: viral causes. Endocrinol Metab Clin North Am 33:27–44, viii
Iwasaki T, Monma N, Satodate R, Kawana R, Kurata T (1985) An immunofluorescent study of generalized Coxsackie virus B3 infection in a newborn infant. Acta Pathol Jpn 35:741–748
Jun HS, Kang Y, Notkins AL, Yoon JW (1997) Gain or loss of diabetogenicity resulting from a single point mutation in recombinant encephalomyocarditis virus. J Virol 71:9782–9785
Kawashima H, Ihara T, Ioi H, Oana S, Sato S, Kato N, Takami T, Kashiwagi Y, Takekuma K, Hoshika A, Mori T (2004) Enterovirus-related type 1 diabetes mellitus and antibodies to glutamic acid decarboxylase in Japan. J Infect 49:147–151
Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA (2006) Enterovirus surveillance—United States, 1970–2005. MMWR Surveill Summ 55:1–20
Kim KS, Tracy S, Tapprich W, Bailey J, Lee CK, Kim K, Barry WH, Chapman NM (2005) 5′-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J Virol 79:7024–7041
Kruppenbacher JP, Mertens T, Muntefering H, Eggers HJ (1985) Encephalomyocarditis virus and diabetes mellitus: studies on virus mutants in susceptible and non-susceptible mice. J Gen Virol 66(Pt 4):727–732
Lonnrot M, Korpela K, Knip M, Ilonen J, Simell O, Korhonen S, Savola K, Muona P, Simell T, Koskela P, Hyoty H (2000) Enterovirus infection as a risk factor for beta-cell autoimmunity in a prospectively observed birth cohort: the Finnish Diabetes Prediction and Prevention Study. Diabetes 49:1314–1318
Lonnrot M, Salminen K, Knip M, Savola K, Kulmala P, Leinikki P, Hyypia T, Akerblom HK, Hyoty H (2000) Enterovirus RNA in serum is a risk factor for beta-cell autoimmunity and clinical type 1 diabetes: a prospective study. Childhood Diabetes in Finland (DiMe) Study Group. J Med Virol 61:214–220
Makela M, Vaarala O, Hermann R, Salminen K, Vahlberg T, Veijola R, Hyoty H, Knip M, Simell O, Ilonen J (2006) Enteral virus infections in early childhood and an enhanced type 1 diabetes-associated antibody response to dietary insulin. J Autoimmun 27:54–61
Moya-Suri V, Schlosser M, Zimmermann K, Rjasanowski I, Gurtler L, Mentel R (2005) Enterovirus RNA sequences in sera of schoolchildren in the general population and their association with type 1-diabetes-associated autoantibodies. J Med Microbiol 54:879–883
Nairn C, Galbraith DN, Taylor KW, Clements GB (1999) Enterovirus variants in the serum of children at the onset of Type 1 diabetes mellitus. Diabet Med 16:509–513
Nejentsev S, Walker N, Riches D, Egholm M, Todd JA (2009) Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324:387–389
Oikarinen M, Tauriainen S, Honkanen T, Oikarinen S, Vuori K, Kaukinen K, Rantala I, Maki M, Hyoty H (2008) Detection of enteroviruses in the intestine of type 1 diabetic patients. Clin Exp Immunol 151:71–75
Oikarinen M, Tauriainen S, Honkanen T, Vuori K, Karhunen P, Vasama-Nolvi C, Oikarinen S, Verbeke C, Blair GE, Rantala I, Ilonen J, Simell O, Knip M, Hyoty H (2008) Analysis of pancreas tissue in a child positive for islet cell antibodies. Diabetologia 51:1796–1802
Parkkonen P, Hyoty H, Koskinen L, Leinikki P (1992) Mumps virus infects beta cells in human fetal islet cell cultures upregulating the expression of HLA class I molecules. Diabetologia 35:63–69
Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG (2009) The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 52:1143–1151
Richer MJ, Horwitz MS (2009) Coxsackievirus infection as an environmental factor in the etiology of type 1 diabetes. Autoimmun Rev 8:611–615
Roivainen M, Ylipaasto P, Savolainen C, Galama J, Hovi T, Otonkoski T (2002) Functional impairment and killing of human beta cells by enteroviruses: the capacity is shared by a wide range of serotypes, but the extent is a characteristic of individual virus strains. Diabetologia 45:693–702
Ross ME, Onodera T, Brown KS, Notkins AL (1976) Virus-induced diabetes mellitus. IV. Genetic and environmental factors influencing the development of diabetes after infection with the M variant of encephalomyocarditis virus. Diabetes 25:190–197
Sadeharju K, Lonnrot M, Kimpimaki T, Savola K, Erkkila S, Kalliokoski T, Savolainen P, Koskela P, Ilonen J, Simell O, Knip M, Hyoty H (2001) Enterovirus antibody levels during the first two years of life in prediabetic autoantibody-positive children. Diabetologia 44:818–823
Sadeharju K, Hamalainen AM, Knip M, Lonnrot M, Koskela P, Virtanen SM, Ilonen J, Akerblom HK, Hyoty H (2003) Enterovirus infections as a risk factor for type I diabetes: virus analyses in a dietary intervention trial. Clin Exp Immunol 132:271–277
Sadeharju K, Knip M, Hiltunen M, Akerblom HK, Hyoty H (2003) The HLA-DR phenotype modulates the humoral immune response to enterovirus antigens. Diabetologia 46:1100–1105
Sadeharju K, Knip M, Virtanen SM, Savilahti E, Tauriainen S, Koskela P, Akerblom HK, Hyoty H (2007) Maternal antibodies in breast milk protect the child from enterovirus infections. Pediatrics 119:941–946
Salminen K, Sadeharju K, Lonnrot M, Vahasalo P, Kupila A, Korhonen S, Ilonen J, Simell O, Knip M, Hyoty H (2003) Enterovirus infections are associated with the induction of beta-cell autoimmunity in a prospective birth cohort study. J Med Virol 69:91–98
Salminen KK, Vuorinen T, Oikarinen S, Helminen M, Simell S, Knip M, Ilonen J, Simell O, Hyoty H (2004) Isolation of enterovirus strains from children with preclinical Type 1 diabetes. Diabet Med 21:156–164
Sauter P, Hober D (2009) Mechanisms and results of the antibody-dependent enhancement of viral infections and role in the pathogenesis of coxsackievirus B-induced diseases. Microbes Infect 11:443–451
Sayama K, Imagawa A, Okita K, Uno S, Moriwaki M, Kozawa J, Iwahashi H, Yamagata K, Tamura S, Matsuzawa Y, Hanafusa T, Miyagawa J, Shimomura I (2005) Pancreatic beta and alpha cells are both decreased in patients with fulminant type 1 diabetes: a morphometrical assessment. Diabetologia 48:1560–1564
Seiskari T, Kondrashova A, Viskari H, Kaila M, Haapala AM, Aittoniemi J, Virta M, Hurme M, Uibo R, Knip M, Hyoty H (2007) Allergic sensitization and microbial load—a comparison between Finland and Russian Karelia. Clin Exp Immunol 148:47–52
Shibasaki S, Imagawa A, Tauriainen S, Iino M, Oikarinen M, Abiru H, Tamaki K, Seino H, Nishi K, Takase I, Okada Y, Uno S, Murase-Mishiba Y, Terasaki J, Makino H, Shimomura I, Hyoty H, Hanafusa T (2009) Expression of toll-like receptors in the pancreas of recent-onset fulminant type 1 diabetes. Endocr J
Shigemoto T, Kageyama M, Hirai R, Zheng J, Yoneyama M, Fujita T (2009) Identification of loss of function mutations in human genes encoding RIG-I and MDA5: implications for resistance to type I diabetes. J Biol Chem 284:13348–13354
Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, Guja C, Ionescu-Tirgoviste C, Widmer B, Dunger DB, Savage DA, Walker NM, Clayton DG, Todd JA (2006) A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet 38:617–619
Tanaka S, Nishida Y, Aida K, Maruyama T, Shimada A, Suzuki M, Shimura H, Takizawa S, Takahashi M, Akiyama D, Arai-Yamashita S, Furuya F, Kawaguchi A, Kaneshige M, Katoh R, Endo T, Kobayashi T (2009) Enterovirus infection, CXC chemokine ligand 10 (CXCL10), and CXCR3 circuit: a mechanism of accelerated beta-cell failure in fulminant type 1 diabetes. Diabetes 58:2285–2291
Tauriainen S, Oikarinen M, Keim J, Oikarinen S, Hyöty H, group ns (2009) Detection of enterovirus in pancreatic tissues of cadaver organ donors—results from nPOD study. In: Annual meeting of Immunology of Diabetes Society, Malmö, Sweden
Tracy S, Drescher KM (2007) Coxsackievirus infections and NOD mice: relevant models of protection from, and induction of, type 1 diabetes. Ann N Y Acad Sci 1103:143–151
Viskari H, Ludvigsson J, Uibo R, Salur L, Marciulionyte D, Hermann R, Soltesz G, Fuchtenbusch M, Ziegler AG, Kondrashova A, Romanov A, Knip M, Hyoty H (2004) Relationship between the incidence of type 1 diabetes and enterovirus infections in different European populations: results from the EPIVIR project. J Med Virol 72:610–617
Viskari H, Ludvigsson J, Uibo R, Salur L, Marciulionyte D, Hermann R, Soltesz G, Fuchtenbusch M, Ziegler AG, Kondrashova A, Romanov A, Kaplan B, Laron Z, Koskela P, Vesikari T, Huhtala H, Knip M, Hyoty H (2005) Relationship between the incidence of type 1 diabetes and maternal enterovirus antibodies: time trends and geographical variation. Diabetologia 48:1280–1287
Viskari HR, Koskela P, Lonnrot M, Luonuansuu S, Reunanen A, Baer M, Hyoty H (2000) Can enterovirus infections explain the increasing incidence of type 1 diabetes? Diabetes Care 23:414–416
Viskari HR, Roivainen M, Reunanen A, Pitkaniemi J, Sadeharju K, Koskela P, Hovi T, Leinikki P, Vilja P, Tuomilehto J, Hyoty H (2002) Maternal first-trimester enterovirus infection and future risk of type 1 diabetes in the exposed fetus. Diabetes 51:2568–2571
von Herrath M (2009) Diabetes: a virus-gene collaboration. Nature 459:518–519
Woodruff JF (1980) Viral myocarditis. A review. Am J Pathol 101:425–484
Yin H, Berg AK, Tuvemo T, Frisk G (2002) Enterovirus RNA is found in peripheral blood mononuclear cells in a majority of type 1 diabetic children at onset. Diabetes 51:1964–1971
Yin H, Berg AK, Westman J, Hellerstrom C, Frisk G (2002) Complete nucleotide sequence of a Coxsackievirus B-4 strain capable of establishing persistent infection in human pancreatic islet cells: effects on insulin release, proinsulin synthesis, and cell morphology. J Med Virol 68:544–557
Ylipaasto P, Klingel K, Lindberg AM, Otonkoski T, Kandolf R, Hovi T, Roivainen M (2004) Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia 47:225–239
Yoon JW, Austin M, Onodera T, Notkins AL (1979) Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med 300:1173–1179
Yoon JW, Notkins AL (1983) Virus-induced diabetes in mice. Metabolism 32:37–40
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is published as part of the Special Issue on Immunopathology of the pancreas in type 1 diabetes.
Rights and permissions
About this article
Cite this article
Tauriainen, S., Oikarinen, S., Oikarinen, M. et al. Enteroviruses in the pathogenesis of type 1 diabetes. Semin Immunopathol 33, 45–55 (2011). https://doi.org/10.1007/s00281-010-0207-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-010-0207-y