
Abstract
Membranous nephropathy, a disease characterized by an accumulation of immune deposits on the outer aspect of the glomerular basement membrane, is the most common cause of idiopathic nephrotic syndrome in white adults. In the rat model of Heymann nephritis, the target antigen of antibodies is megalin, a multiligand receptor expressed at the podocyte cell surface. This review summarizes key findings provided by this experimental model and by our discovery of neutral endopeptidase being the alloantigen involved in neonatal cases of membranous nephropathy. We discuss the role of alloimmunization as a new mechanism of renal disease and the approach that we use to identify new podocyte antigens. We also summarize current knowledge on the mechanism of proteinuria, with special emphasis on the role of complement. In conclusion, substantial progresses have been made in understanding molecular mechanisms of membranous nephropathy, which should lead to novel therapeutic approaches.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Membranous nephropathy is the most common cause of idiopathic nephrotic syndrome in white adults, accounting for about 20% of cases. Although spontaneous remission of nephrotic syndrome occurs in about a third of patients, membranous nephropathy ends for about 40% of patients in end-stage renal failure after 10 years [1, 2]. Eighty percent of cases are classified as idiopathic, to conceal our ignorance about causes, while about 20% present with associate clinical conditions including infections, autoimmune diseases, cancers, and are thus classified as having secondary disease. Treatment of membranous nephropathy is often disappointing [3, 4]. This is due in part to heterogeneity of the disease and lack of reliable biomarkers because of ignorance of the target antigen(s) and nephritogenic antibodies. Strategies to target B lymphocytes with anti-CD20 antibody [5] and to inhibit complement [6] are steps in the right direction, but more specific concept-driven therapies are urgently needed.
Membranous nephropathy is characterized by an accumulation of immune deposits on the outer aspect of the glomerular basement membrane, which causes a membrane-like thickening (Fig. 1). The immune deposits consist of IgG, with a predominance of IgG4 [7, 8], so far unidentified antigens, and the membrane attack complex of complement C5b-9 (MAC). Functional impairment of the glomerulus causing proteinuria results from the formation of subepithelial immune deposits and complement activation. The key to a specific hypothesis-driven therapy is the understanding of the development of immune deposits, which first requires identification of the pathogenic antigen(s), and of the ensuing events mediated by C5b-9. This review focuses on the molecular pathomechanisms of membranous nephropathy with particular emphasis on the antigenic targets of nephritogenic antibodies.

Granular immune deposits revealed by immunofluorescence with an anti-IgG antibody (a) and by electron microscopy (b) in a patient with membranous nephropathy. Note that deposits are discontinuous and exclusively located on the outer space of the glomerular basement membrane. Podocyte foot processes appear flattened by electron microscopy in this patient with abundant proteinuria. US Urinary space, GBM glomerular basement membrane, Pod podocyte
Heymann nephritis: the rat experimental model of autoimmune membranous nephropathy
We have learned a great deal about idiopathic membranous nephropathy from Heymann nephritis, which provided the bases of molecular and kinetic concepts of immune deposit formation and glomerular capillary wall injury. The active model of Heymann nephritis is induced by immunization of Lewis rats with preparations of brush-border proteins [9].
Initial studies of this model suggested that the subepithelial deposits resulted from glomerular trapping of circulating immune complexes formed by circulating brush-border-related antigens and the corresponding antibodies. This hypothesis was based on the observation that the glomerular disease was induced by fractions of membrane prepared from rat renal brush border, not from glomerular extracts.
Subsequently, the development of the model of passive Heymann nephritis in rats that received an injection of rabbit anti-rat brush-border antibodies led to the suggestion that subepithelial immune deposits could be formed without the intervention of circulating immune complexes. Van Damme et al. [10] and Couser et al. [11], using ex vivo and isolated perfused kidney systems, further demonstrated that anti-brush-border antibodies could bind glomeruli in the absence of circulating brush-border-related antigen, which provided the proof of principle that immune complex formation occurred in situ. Definitive evidence establishing the role of in situ immune complex formation in the glomerular capillary wall required identification of the antigen moiety.
Identification of megalin, a new rat podocyte protein
The autoantigenic target in the rat disease was identified by Kerjaschki and Farquhar [12, 13] in the early 1980s as the podocyte membrane protein now called megalin. The polyspecific receptor megalin, a member of the low-density lipoprotein-receptor superfamily, is expressed with clathrin at the sole of podocyte foot processes (where immune complexes are formed). The system was dissected on a molecular level to the precise amino acid sequence of pathogenic epitopes (see below). The continued growth of immune deposits seems to require the de novo synthesis by the podocytes of new molecules of megalin, which are assumed to be delivered via vesicles that eventually fuse with the cell membrane at the base of the foot processes [14]. These findings provided the first evidence that podocytes actively contribute to the formation of glomerular immune deposits in membranous nephropathy.
Antibodies to receptor-associated protein, a 39-kDa protein which acts as a chaperone for and binds to megalin [15], were also detected in rats with Heymann nephritis, and passive Heymann nephritis could be induced by monospecific antibodies against receptor-associated protein. However, the rats did not develop proteinuria, and ID were cleared within a few weeks from the kidneys of rats injected with anti-receptor-associated protein antiserum [16]. Furthermore, receptor-associated protein by itself could not induce active Heymann nephritis. These results indicate a clear divergence in pathogenic potential of megalin and receptor-associated protein, which may be related to the fact that megalin is an integral membrane protein, whereas receptor-associated protein is not bound to the podocyte membrane.
Towards identification of nephritogenic megalin epitopes
Numerous attempts have been made to dissect the megalin system on a molecular level, which is a prerequisite for specific immunointervention. Cloning of megalin gene in 1994 [17] revealed that megalin is an ∼4,600-amino acid (aa) transmembrane protein with a molecular weight of ∼600 kDa [15]. Given the large size of megalin, the pinpoint by Raychowdhury et al. [18] of a 137-aa fragment (aa 1,114 to 1,250) in the second ligand-binding domain as a pathogenic epitope recognized by antibodies eluted from the glomeruli of rats with active Heymann nephritis represented a major breakthrough (Fig. 2). Saito et al. [19] narrowed the epitope to the fifth ligand-binding repeat consisting of 46 aa (aa 1,160 to 1,205). In fact, all four putative megalin ligand-binding domains actually contain pathogenic epitopes capable of inducing passive Heymann nephritis (i.e., granular subepithelial immune deposits) [20]. However, proteinuria was not reported in either of these models. Therefore, the finding by Makker and coworkers [21] that a 60-kDa N-terminal fragment (nM60) encompassing aa 1 to 563 could induce full-blown active Heymann nephritis was a significant feat. Now, by successive C-terminal truncations, Tramontano et al. [22] have further narrowed the pathogenic epitopes to aa 157–236 in the first ligand-binding domain. Three additional findings are of interest. First, full immunogenic activity required expression of the fragments in insect cells, suggesting that posttranslational modifications and/or conformational determinants are essential for the pathogenic potential [22, 23]. Second, lymph node cell proliferation assays indicated that the pathogenic epitopes could elicit T cell responses. Third, levels of B cell responses in rats immunized with different fragments did not correlate with severity of the disease, which suggests that qualitative differences in the immune response including epitope specificity and isotype distribution are of paramount importance.

The structure of megalin indicating the regions of megalin containing pathogenic epitopes in membranous nephropathy. Megalin is a 4,600-aa transmembrane protein. The extracellular domain contains four cystein-rich clusters of low-density lipoprotein-receptor type A repeats which constitute the ligand-binding domains and are separated and followed by 17 epidermal growth factor EGF-type repeats and eight spacer regions that contain YWTD repeats. Molecular determinants on the region composed of residues 157–236 (red bars in the scheme) are critical for expression of the full disease. This fragment initiates a primary immune response and subsequently triggers epitope spreading to the other ligand-binding domains (black bar in the scheme). Epitope spreading enhances polyvalent cross-linkage leading to the formation of more stable deposits and complement fixation
Makker’s group further showed that, after immunization with L6 (the 236-residue N-terminal megalin fragment), a process of intramolecular epitope spreading occurred, as defined by appearance of antibodies and lymph node cell reactivity directed against additional megalin epitopes not included in the immunizing fragment, but expressed by the ligand-binding domains [24]. The onset of proteinuria correlated with the appearance of anti-ligand-binding domain antibodies rather than with the antibodies induced by L6. This indicates that induction of a nephritogenic response is a complex process which may require multivalent interactions with the target antigen [14], as well as involvement of appropriate Ig isotypes.
Although considerable insight in the mechanisms of immune complex formation and nephritogenic potential has been provided by studies of Heymann nephritis, megalin cannot be taken responsible for human membranous nephropathy because it has not been found in human glomeruli or podocytes, nor it has been detected in subepithelial immune deposits in patients with membranous nephropathy. In fact, the rat is the only species where megalin is detected in glomeruli, although megalin is found in the brush border in all species as yet studied, including humans.
Enzymatic antigens involved in the formation of subepithelial immune deposits
The enzymes dipeptidyl peptidase IV (DPPIV) [25], neutral endopeptidase [26], and aminopeptidase A [27] were shown to serve as target antigens for circulating antibodies in rats, rabbit, and mice, respectively. At variance with megalin, DPPIV is evenly distributed on the membrane of podocytes, is further expressed on endothelial cells lining the glomerular capillary wall, and assumes a wide extrarenal distribution including transport epithelia, capillary endothelia, and the vast majority of normal lymphocytes [28, 29]. The kinetics of immune deposits observed in the rat glomerulus after intravenous injection of anti-DPPIV monoclonal or polyclonal antibody sharply contrast with those noticed in rats given anti-megalin antibody [25]. Glomerular binding is maximum 4 h after injection, but from then on decreases dramatically to be absent or very weak 72 h later. In the mouse, the kinetics of the heterologous phase after injection of anti-DPPIV antibody bear close similarity to those reported in the rat, but the amount of antibody remaining in the glomerulus is sufficient to induce the development of an autologous phase that causes membranous nephropathy [30, 31]. DPPIV–anti-DPPIV immune complexes formed at the surface of glomerular endothelial cells may be shed in the capillary lumen, partly dissociate, then be filtered through the glomerular capillary wall, and finally reassociate on the outer aspect of the glomerular basement membrane. This pathophysiological scenario does occur in a model of membranous nephropathy induced in the rabbit by polyclonal antibodies to angiotensin-converting enzyme, which is expressed by glomerular endothelial cells, but not by rabbit’s podocytes [32].
The second antigen, neutral endopeptidase, has the same distribution in the rabbit and human kidneys as DPPIV, whereas in the rat, it is found on cells of the Bowman’s capsule and in the distal segment of the proximal tubule (pars recta) [33]. Glomerular deposits observed after injection in rabbit of monoclonal antibody to neutral endopeptidase are particularly transient. Their almost complete disappearance within 24 h coincides with the appearance of the antibody on the brush border of some proximal convoluted tubules, as noted in the rat with anti-DPPIV.
Because both DPPIV and neutral endopeptidase are expressed on the human podocyte, we hypothesized about 20 years ago that those two enzymatic antigens might play some role in the pathogenesis of membranous nephropathy in humans [26].
Fetomaternal alloimmune glomerulopathies: the human counterpart of passive Heymann nephritis
After 20 years of research since the discovery of megalin, we identified a human counterpart to the Heymann nephritis antigen in a patient with neonatal membranous nephropathy [34]. The male infant who was born at 38 weeks of gestation presented with oligoanuria, massive proteinuria, and respiratory distress on the first day of life. His parents were unrelated, healthy individuals without a family history of renal or autoimmune disease. The mother, aged 24, had had a miscarriage at 14 weeks of gestation 2 months before this pregnancy. Her blood pressure, urinalysis, and serum creatinine concentration were normal throughout and after the pregnancy, and she took no medications. However, antenatal echography showed oligohydramnios and enlarged fetal kidneys from the 34th week of gestation. Her levels of antineutrophil cytoplasmic antibodies, antinuclear and anti-DNA antibodies, and complement were normal.
Identification of neutral endopeptidase as the target antigen of nephritogenic antibodies
Because of the early development of membranous nephropathy in this infant, we suspected pregnancy-induced immunization of the mother with transplacental passage of nephritogenic antibodies. This hypothesis was first tested by indirect immunofluorescence examination of normal human kidney sections. A serum sample obtained 9 months before pregnancy (7 months before the miscarriage) was negative. Serum samples obtained at 3 months of gestation and after delivery showed reactivity on the glomerular capillary walls and the brush border in all kidney biopsy specimens, as did the serum obtained from the infant 13 days after birth. No reactivity was detected in the infant’s serum 40 days after birth, which confirmed that “anti-kidney” antibodies circulating in the infant’s serum were of maternal origin [34].
The nature of the target antigen was suspected by indirect immunofluorescence examination of rabbit and rat kidney sections incubated with the mother’s or the infant’s antibody. The same pattern as in human kidneys was observed in the rabbit, whereas in the rat, staining was restricted to the cells of Bowman’s capsule and to the brush border of deep cortical segments of the proximal tubule. We had previously observed similar interspecies differences with antineutral endopeptidase antibodies, whereas distribution of DPPIV is not species dependent [33]. The mother’s IgG antibody and the infant’s IgG antibody recognized by Western blotting a single antigen of approximately 90 kDa in protein extracts from rat brush border, rabbit kidney cortex, and cultured human podocytes. This antigen had the same electrophoretic mobility as neutral endopeptidase. Furthermore, neutral endopeptidase antigen and enzymatic activity were specifically immunoprecipitated from rat brush border with the mother’s IgG [34].
The antineutral endopeptidase antibodies produced by the mother, which were found in the infant’s serum 13 days after birth, were most likely responsible for the infant’s membranous nephropathy, given that the injection of rabbits with the serum IgG fraction from the mother induced intraglomerular deposits and proteinuria, whereas injection with the IgG fraction from the father did not. Furthermore, neutral endopeptidase was localized by confocal microscopy in immune deposits together with the MAC of complement, both in the infant and in the rabbits injected with the mother’s IgG.
Since the description of the index case, we have identified two other families, one in The Netherlands, the other one in Belgium but of Moroccan origin, with at least one infant born with membranous nephropathy and the same mechanism of disease [35]. Interestingly, the Dutch case was reported in 1990; at that time, the mother’s serum had been tested for the presence of anti-DPPIV antibodies, but not for that of antineutral endopeptidase antibodies [36].
Mechanisms of neutral endopeptidase–antineutral endopeptidase immune complex deposition in the infants’ glomeruli
The four cases of antenatal membranous nephropathy because of transplacental transfer of antineutral endopeptidase antibodies led us to revisit the concept of in situ formed vs circulating preformed immune complexes, which has remained a debated issue for the last 30–40 years or so. It is most likely that, in these cases, immune complexes were predominantly formed in situ at the sole of podocyte foot process where neutral endopeptidase is expressed [37] (Fig. 3a). Neutral endopeptidase is expressed in a diffuse pattern on the membrane of podocytes, as is angiotensin-converting enzyme on the plasma membrane of mature oocytes [38]. In vivo interaction of angiotensin-converting enzyme with divalent antibodies induces the formation of granular immune deposits through a mechanism of “patching” and “shedding” of immune complexes [38]. A similar mechanism may be implicated in the formation of immune deposits in the infants’ glomeruli. One can speculate that the immune complexes that are shed from the foot processes are sequestered between the lamina rara externa of the glomerular capillary wall and the podocytes’ slit diaphragms, whereas those that are shed from the podocyte cell bodies are excreted in the infant’s urine.

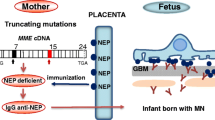
Mechanisms of immune complex formation and podocyte injury in alloimmune neonatal membranous nephropathy. a In situ formation of immune deposits in neonatal membranous nephropathy. Neutral endopeptidase (blue dots) serves as pathogenic antigen in the podocyte’s cell membrane. Antibodies to this protein originate in women who lack neutral endopeptidase epitopes because of truncating mutations. Antiendopeptidase antibody is transported across placenta and causes formation of immune complexes at podocyte membranes, similar to those observed in experimental Heymann nephritis. It is likely that as for megalin (the antigen of Heymann nephritis), neutral endopeptidase–antineutral endopeptidase immune complexes formed on the podocyte membrane are then shed and rapidly immobilized in the glomerular basement membrane, thus preventing clearing of complexes by endocytosis in the podocyte. b Schematic description of the cellular mechanisms that lead to proteinuria in membranous nephropathy. C5b-9 formation on the membrane of podocytes triggers various intracellular events, including production of ROS and proteases, and cytoskeletal changes. These result in degradation of glomerular basement membrane and redistribution of proteins that compose the slit diaphragm, eventually leading to development of protein leakage into the Bowman’s space (left). In addition, C5b-9 attack leads to podocytopenia through apoptosis, lack of proliferation resulting from complement induced DNA damage, and podocyte detachment (right)
However, transient low levels of circulating immune complexes were detected in the infant’s serum. The immune complexes isolated from the serum sample contained neutral endopeptidase [34]. Their contribution to the formation of subepithelial immune deposits is uncertain because levels of circulating immune complexes were low, manifestations of serum sickness were absent, and subendothelial and mesangial immune deposits were not seen. The two mechanisms of immune complex formation (in situ vs preformed) are not mutually exclusive.
Alloimmunization: a novel mechanism of renal disease
Mechanisms of the immunization against neutral endopeptidase in the infant’s mother
Because the first mother reported [34] had no apparent renal abnormalities despite high serum titers of antineutral endopeptidase antibody, we hypothesized that she might be deficient in neutral endopeptidase and analyzed neutral endopeptidase expression in granulocytes from both parents. Fluorescence-activated cell sorter analysis of the mother’s granulocytes showed no neutral endopeptidase at the cell membrane. Cell extracts prepared from maternal granulocytes failed to react with either monoclonal or polyclonal antibodies against neutral endopeptidase after Western blotting. Moreover, the mother’s serum reacted with the father’s granulocytes but not with her own granulocytes, suggesting an alloimmunization process. Alloimmunization in the mother most likely occurred at the time of her miscarriage, given that a plasma sample obtained earlier did not show antineutral endopeptidase antibodies [34]. At that time, the mother’s immune system was massively exposed to neutral endopeptidase antigen expressed by syncytiotrophoblasts and fetal cells.
Identification of mutations in the mothers’ neutral endopeptidase gene
We found that the four other antineutral-endopeptidase-immunized mothers from the Dutch and Moroccan families were also neutral endopeptidase deficient, which led us to search for mutations in the MME gene for neutral endopeptidase [35]. The MME gene is composed of 24 exons. Exons 3 to 24 encode a 749-aa protein that consists of a short cytoplasmic domain, a transmembrane domain, and a large extracellular moiety with a zinc-binding motif required for enzymatic activity. We identified two truncating mutations in these families (Fig. 4). The first mutation, located in exon 7, is a cytosine deletion at position 466 that results in a frameshift and premature termination codon at codon 169. The second mutation, located in exon 15, is a single-base nonsense mutation (1342C→T) that generates a stop codon at position 448. The Portuguese mother is a compound heterozygote who inherited one mutant allele from each parent, whereas the Dutch and Moroccan mothers were homozygous for the same deletion mutation 466delC and inherited the mutant allele from their heterozygous parents (Fig. 4).

Pedigrees and distribution of IgG subclasses in families with FMAIG. a Pedigrees of the three families. Roman numerals indicate generations in the respective families. Families are from Portugal (P), The Netherlands (N), and Morocco (M). Black Segregation of mutation in exon 7, red segregation of mutation in exon 15. Blue asterisk indicates the mother with antineutral endopeptidase antibodies; blue plus symbol indicates the children born with membranous nephropathy. b Distribution of neutral-endopeptidase-specific IgG subclasses in the sera of mothers determined by Western blot analysis. Note that mother IIM8 showed a low titer of antineutral endopeptidase antibodies, which were exclusively of the IgG4 subclass. Her children had no overt manifestation of neonatal renal disease. In contrast, the four remaining mothers had a high titer of antineutral endopeptidase, which belonged to the IgG1 and IgG4 subclasses. Their children were born with membranous nephropathy
Theoretically, mutations in exon 7 and exon 15 lead to highly truncated proteins of 168 and 450 aa, respectively, devoid of enzymatic activity (the zinc-binding motif is encoded by exon 19). However, we failed to detect truncated proteins in the mothers’ granulocytes and urine samples [35]. These findings indicate that the mutated MME gene is knocked out functionally probably because of decreased stability of the mutated mRNA or protein.
Despite the absence of neutral endopeptidase protein in the five mothers and in a male individual, these individuals, aged 16 to 42, were healthy (as were heterozygous family members except for the neonates who were born with membranous nephropathy). By contrast with MME null mice [39], they had normal blood pressure, renal functional tests, and lymphocyte phenotype and function [35]. The lack of apparent consequence of neutral endopeptidase deficiency can be partly explained by redundancy of the enzyme activity [40, 41].
We, thus, have characterized a novel fetomaternal disease in which a genetic defect in the mother leads to the development of membranous nephropathy in her fetus. Currently, Rhesus incompatibility is the paradigm of fetomaternal diseases because of alloimmunization, and such diseases have been described only for red blood cells and platelets. Our findings raise the possibility that truncating mutations in other podocyte antigens, asymptomatic for the carrier mother, could lead to fetomaternal alloimmune glomerulopathies (FMAIG). Similarly, immunization against allovariants of proteins expressed by placental cells in the mother and by glomerular cells in the fetus might cause neonatal renal disease.
Alloimmune membranous nephropathy in the renal graft
The discovery of alloimmune neonatal membranous nephropathy induced by antineutral endopeptidase antibodies might also shed new light on the pathogenesis of de novo membranous nephropathy which develops after renal transplantation [42]. Indeed, analogies can be drawn between the pregnant mother and the graft recipient on the one hand, and the fetus (and the placenta) and the kidney donor on the other. Because neutral endopeptidase deficiency is asymptomatic in humans, neutral-endopeptidase-deficient graft recipients are not identified before transplant. These individuals are most likely to raise an antineutral endopeptidase alloimmune response when their immune system is exposed to neutral endopeptidase in the donor kidney. Other kidney antigens might elicit a similar response if the recipient is genetically deficient or expresses an allovariant.
Alloimmune membranous nephropathy in the adult native kidney: from graft-versus-host disease to spontaneously occurring microchimerism
The view that alloimmune nephropathies occur in adult native kidneys is supported by observations made in patients receiving a bone marrow transplant or allogenic blood stem cells, and those suffering from graft-versus-host disease (GVHD) [43–47]. To our knowledge, glomerulopathy subsequent to hematopoietic stem cell transplantation has been reported in less than 30 cases in the English literature; however, incidence might increase with new chemotherapy regimens, as has occurred recently. Membranous nephropathy is by far the most common histologic lesion [48] accounting for more than 30% of cases. The pathogenetic mechanism of glomerulopathy after hematopoietic stem cell transplantation remains unclear.
Existing GVHD is considered to be a key event in development of renal disease of this nature. GVHD mimics the biological and clinical features of systemic lupus erythematosus or other immune-complex-related disorders. In experimental models of GVHD, glomerular injury has been observed in association with immune complexes [49]. Circulating immune complexes are too large to traverse the glomerular capillary wall. They might partially dissociate on the endothelial side of the glomerular capillary wall, before crossing and reassembling on the epithelial side. Alternatively, podocyte-associated proteins might serve as targets for the circulating alloimmune antibodies that are directed against a podocyte antigen expressed in the recipient but absent from the donor, or against an allovariant.
GVHD is a typical example of acquired microchimerism that fulfills the classical definition; that is, presence of a small population of cells (or DNA) in one individual that is derived from another genetically distinct individual. In addition to iatrogenic chimerism occurring as a consequence of hematopoietic cell transplantation, organ transplantation, and blood transfusion, it has recently been found that cell trafficking between mother and fetus during pregnancy can result in long-term persistence of fetal cells (fetal cell microchimerism) in the mother [50], and of maternal cells in her progeny (maternal cell microchimerism) [51]. Although not yet proven, fetal cell microchimerism is presumed to persist after miscarriage and abortion. Concerns about the histocompatibility of these fetal or maternal cells have raised questions about the long-term consequences of an immune response on the mother’s and child’s health, respectively.
Maternal cell microchimerism has been investigated in systemic sclerosis, dermatomyositis, and neonatal lupus [52]. On the other hand, the possible connection between fetal cell microchimerism and autoimmune disease has received much attention because autoimmune disease occurs predominantly in women, and after the peak of childbearing years. Some autoimmune diseases also have features of chronic GVHD. The question all studies have raised is whether fetal cells, once established in women after pregnancy, can trigger an alloimmune response and initiate an “autoimmune” disease [53]. Systemic sclerosis was the first autoimmune disease to be studied. We favor the hypothesis that microchimerism has a role in some renal diseases, particularly membranous nephropathy. This hypothesis is supported by the occurrence of membranous nephropathy in GVHD (as discussed above), and production of autoantibodies against nephrin in an experimental model [54]. Microchimerism might thus trigger production of antibodies against podocyte antigens by mechanisms that remain to be elucidated.
Which target antigens for adult membranous nephropathy?
Is neutral endopeptidase still a candidate?
We have investigated the outcome of antenatal membranous nephropathy in the four infants who were born to neutral-endopeptidase-immunized mothers. All infants showed a rapid improvement of renal failure and the nephrotic syndrome. However, children IV P1 and III M1 (Fig. 4) showed persistent albuminuria. Patient III N1, now 20 years old, is of particular clinical interest because of the postponed development of severe chronic renal failure with nephrotic-range proteinuria. Although we could not undertake a second kidney biopsy in the oldest patient, current renal manifestations are likely to result from an aged membranous nephropathy combined with the delayed consequences of immunologically mediated antenatal nephron loss. Deposition of IgG produced by infants to idiotypes or allotypes on the maternal IgG could contribute to later progression of the disease. These observations suggest that antineutral-endopeptidase-induced antenatal renal disease might account for “idiopathic” membranous nephropathy or chronic renal failure detected during adolescence or early adulthood.
As yet, we have failed to find neutral endopeptidase in the subepithelial immune deposits in patients with “idiopathic” membranous nephropathy. This does not rule out a role for neutral endopeptidase in the disease because the initiating antigen may no longer be present in aged immune deposits. However, should antineutral endopeptidase antibody be produced, they would bind to neutral endopeptidase that is heavily expressed on granulocytes; therefore, they would not be available for their glomerular target antigen. Conversely, one can also hypothesize that neutral endopeptidase–antineutral endopeptidase immune complexes formed on the surface of granulocytes are shed in the serum, where they partially dissociate, allowing their components to traverse the basement membrane before they reassociate between the lamina rara externa and the slit diaphragms.
We hypothesize that neutral endopeptidase might mostly serve as an alloantigen in alloimmune conditions including FMAIG, de novo membranous nephropathy in the grafted kidney, and membranous nephropathy occurring after allogenic bone marrow transplantation.
The case of secondary membranous nephropathy
In so-called secondary forms of membranous nephropathy, hepatitis B, hepatitis C, and Helicobacter pylori antigens; tumor antigens; thyroglobulin; and DNA-containing material have been detected in the subepithelial deposits, but there is no real proof that these antigens are pathogenic [55, 56] (Table 1; [57–63]). Because of the increased permeability to proteins of the glomerular capillary wall, they may have been trapped passively between the lamina rara externa and the slit diaphragm as is the case for albumin [64]. Some similarities, such as glomerular deposition of renal tubular epithelial antigens, have been found between experimental Heymann nephritis and individual cases of membranous nephropathy, but the antigens could not be characterized at the molecular level [65–67].
Childhood membranous nephropathy occasionally may be associated with linear or granular deposits of IgG and C3 along the tubular basement membrane. This rare subgroup of patients may have proximal tubule impairment and extrarenal manifestations, including lung hemorrhage, diarrhea because of intestinal villous atrophy or autoimmune enteropathy, and cornea and neurologic symptoms [68]. Antibodies to the 58-kDa tubulointerstitial nephritis (TIN) antigen were reported in 5 of the 11 reported cases [68]. The TIN antigen is a glycoprotein molecule [69] that has the highest expression in the basement membrane of the proximal tubules, whereas it is absent from the glomerular basement membrane and the mesangial matrix [70]. Therefore, the TIN antigen cannot serve as a target glomerular antigen for circulating antibodies. Moreover, careful analysis of the reports strongly suggests that, in childhood membranous nephropathy with antitubular basement membrane nephritis, the glomerular disease is the primary lesion, and the formation of antitubular basement membrane antibodies and their fixation to the tubular basement membrane and the development of the tubulointerstitial disease are secondary phenomena. Because some cases are associated with anti-brush-border antibodies [71], identification of the relevant antigen(s) would be of great value.
The case of idiopathic membranous nephropathy
Most eluates from kidneys of patients with membranous nephropathy do not react with normal kidney [72], which should not be taken as an argument against in situ formation of immune complexes implicating podocyte antigens because eluates were usually obtained in late stage of the nephropathy. At that stage, immune deposits differ significantly from initial immune reactants because the immune deposits may be perpetuated by a secondary anti-idiotype antibody response directed against the original antibody, and also because the composition of immune deposits is continuously being altered by incorporation of passively trapped molecules that have traversed the diseased glomerular capillary wall.
We think that a common denominator to idiopathic and at least some secondary membranous nephropathy is that podocytes and their membrane-associated proteins have a pivotal role in the development of the disease by providing antigenic targets for circulating antibodies for in situ formation of glomerular deposits. We are currently searching for circulating autoantibodies directed to target antigens on human podocytes in sera of patients with membranous nephropathy. To this end, we have developed sensitive assays because the concentration of nephritogenic circulating antibodies is likely to be low, as most of those antibodies may already be deposited in glomeruli. We have recently characterized several profiles of antipodocyte autoantibodies as well as several podocyte antigens by mass spectrometry. Contrary to neutral endopeptidase and megalin, most of those antigens are mostly located in the cytoplasm or cytoskeleton, and some of them are implicated in important metabolic pathways. We do not know yet whether they are involved in the initial phase of the disease as targets for circulating antibodies or whether they are released after podocyte injury induced by complement activation, then triggering a second wave of immunization. The nephritogenic potential of the antibodies specific for those antigens remains to be established.
Fire at the podocyte surface: what causes podocyte injury and proteinuria?
Once immune complexes are deposited in the subepithelial space under the slit diaphragm, a cascade of complex events is triggered, leading to podocyte injury and massive proteinuria (nephrotic syndrome; Fig. 3b). Complement activation certainly plays a major pathogenic role, but other factors including IgG subclass and a direct effect of nephritogenic antibodies should not be dismissed.
Role of complement activation
Complement is a crucial mediator of podocyte injury in experimental membranous nephropathy [73]. Both C6 and C8 are required for the development of proteinuria [74, 75]. We showed that the subepithelial immune deposits in antenatal membranous nephropathy contained heavy deposits of C5b-9 that were colocalized with the neutral endopeptidase antigen and the IgG antibodies [76].
Activation of the classical pathway of complement activation is required to induce the disease. Both in active [77] and passive [73] Heymann nephritis, only the IgG subclasses that are very effective at binding C1q lead to proteinuria. A similar requirement for a classical pathway-activating human IgG isotype is seen in antenatal membranous nephropathy.
However, the unique role of the classical pathway of complement activation is challenged by the fact that most cases of idiopathic human membranous nephropathy have a predominance of IgG4, with less IgG3 and no IgG1 in immune deposits [78, 79]. Furthermore, there is little to no demonstrable C1q and C4 in these deposits [80]. These findings suggest that the alternative pathway of complement activation might be involved. The alternative pathway, which is spontaneously active, is controlled by complement regulatory proteins including membrane complement receptor 1 (Crry in rodents) and decay-accelerating factor that both are expressed at the podocyte surface. Alternative pathway activation and C5b-9 generation on the podocyte in membranous nephropathy could occur because of absent, dysfunctional, or inhibited complement regulatory proteins. There is no evidence that podocyte complement regulatory proteins are targets of autoantibodies in human membranous nephropathy. However, the possibility that CR1 is cleaved by proteases or that it is absent has been investigated by Moll et al. [81], who have shown that the entire protein is absent in glomerular diseases, including membranous nephropathy. Whether absence of CR1 represents an intrinsic or acquired defect and whether this contributes to disease pathogenesis remains to be established [6].
In the rat model, glomerular damage induced by C5b-9 is presumably mediated by the formation by podocytes of reactive oxygen species (ROS) [82]. Although these reactive compounds can directly damage matrix proteins, their effect is further potentiated by local peroxidation of lipids, which can be suppressed by treatment with the scavenger probucol [83]. It is possible that a sequence of complement activation, ROS formation, and lipid peroxidation contributes to glomerular damage and proteinuria [84].
C5b-9, directly or via the production of ROS, can enhance expression by podocytes of matrix metalloproteinase-9 [85, 86], a matrix-degrading enzyme with targeted activity on collagen type IV (a main component of the glomerular basement membrane), and alter nephrin expression [87]. In patients with membranous nephropathy, a more granular pattern or a loss of staining of nephrin was observed [88]. Nephrin is linked to the actin cytoskeleton via nck and CD2AP (review in [89]). C5b-9 formation leads to cytoskeleton changes of podocytes [90] with dissociation of nephrin from the actin cytoskeleton and development of proteinuria [88, 91]. However, the molecular mechanisms whereby C5b-9 induces cytoskeleton changes remain to be established. Cathepsin L was recently identified as a new mediator of proteinuria through cleavage of GTPase dynamin resulting in foot process effacement [92]. Whether cathepsin L is induced by C5b-9 is a timely issue.
Other noxious effects of C5b-9 include an increase in cyclooxygenase-2 and eicosanoid production [93], and in production of laminin and type IV collagen [94]. The effects of C5b-9 on extracellular matrix production, which are likely TGF-β-driven, may be responsible for the spike-like extension of matrix between the immune deposits on the subepithelial aspect of the glomerular basement membrane.
Role of IgG subclass
The glomerular deposition of IgG1 and IgG4 subclasses is a characteristic feature of membranous nephropathy [7, 8]. Idiopathic membranous nephropathy is characterized by a large predominance of IgG4, whereas secondary forms including lupus- and neoplasia-related membranous nephropathy show only weak deposits of IgG4 contrasting with heavy deposits of the other subclasses (Table 2) [95, 96]. Both IgG1 and IgG4 subclasses were found in the diseased infants’ biopsy specimens [35]. However, the expression of the renal disease was variable in the infants who were born to the five mothers who had produced antineutral endopeptidase antibodies. The infants from four mothers presented at birth with renal failure, whereas all four children from mother IIM8 had no overt manifestation of renal disease either at birth or at the most recent follow-up assessment. We found that this mother produced approximately ten times less antineutral endopeptidase antibody than the others, and, perhaps more important, she produced only IgG4 subclass antibodies, whereas the four other mothers produced both IgG1 and IgG4 antineutral endopeptidase antibodies (Fig. 4b). The lack of renal manifestation in the neonates is not explained by deficient transplacental transfer of IgG4 because, at birth, fetal and maternal IgG3 and IgG4 concentrations are normally equal, whereas IgG1 and IgG2 concentrations are higher and lower in the fetus than in the mother, respectively [97]. A more plausible explanation is that IgG subclasses differ in their ability to induce cell injury because they interact differently with complement and Fcγ receptors [98]. By comparison with IgG1, even aggregated IgG4 can weakly activate complement [99], a key mediator of proteinuria in membranous nephropathy as discussed above.
Functional damage induced by antimegalin and antineutral endopeptidase antibodies
In addition to forming immune complexes, antipodocyte antibodies may directly alter podocyte biology. Antimegalin antibodies were shown to inhibit the uptake of lipoproteins that normally bind the multiligand receptor megalin [100]. The accumulation of lipoproteins (apoE and apoB) within the immune deposits together with the production of ROS potentially favors the formation of lipid peroxidation products (see above).
Neutral endopeptidase is involved in the catabolism of a number of regulatory peptides with vasoactive properties, including bradykinin, atriopeptin, and endothelins, and plays an important role in turning off peptide-signaling events at the cell surface. In the human kidney, it is found on podocyte, brush border, and vascular smooth muscle cells [101]. For evaluating a potential effect of antineutral endopeptidase antibodies on enzymatic activity, lysates of human podocytes were preincubated with maternal or paternal IgG. The neutral-endopeptidase-specific activity of podocyte lysates was dose-dependently inhibited by maternal but not paternal IgG. These findings suggest that some of the deleterious effects of antineutral endopeptidase antibody might be mediated by the blockade of neutral endopeptidase enzymatic activity. In the first case reported [34], the infant’s kidney biopsy specimen showed unusually severe arterial lesions without immune deposits and a collapse of glomerular capillary tufts that was suggestive of major renal ischemia during prenatal development. Because the mother’s antibodies inhibited neutral endopeptidase activity, their transplacental passage might increase concentrations of vasoconstrictor peptides, particularly endothelin, in the vascular wall and thus induce the proliferation of vascular smooth muscle cells. The antineutral endopeptidase antibodies might also induce podocyte alterations and proteinuria via their blocking enzyme activity, as previously shown after injection of an anti-aminopeptidase A monoclonal antibody in the mouse [27].
Neutral endopeptidase can also act through direct protein–protein interaction. The neutral endopeptidase cytoplasmic tail might play a key role in providing a scaffold for signaling proteins in the regulation of cell proliferation and organization of the membrane-associated cytoskeleton [102, 103].
However, given the size of subepithelial deposits and the intensity of C5b-9 staining, it is likely that podocyte alterations and increased permeability of the glomerular capillary wall resulted mostly from immune complex formation.
Further studies are needed to establish the role of neutral endopeptidase in podocytes.
Towards antigen (epitope)-driven therapies in patients with membranous nephropathy
Current treatments of patients with membranous nephropathy are entirely empirical, and concept-driven therapies are dramatically lacking. The design of specific therapies for autoimmune diseases is primarily based on induction of specific immune tolerance. This requires ideally identification of the pathogenic epitopes born by the antigen. One way to induce tolerance is mucosal administration of the antigen or immunodominant synthetic peptides. Nasal administration of recombinant NC1 domain of the α3 chain of type IV collagen was shown to induce tolerance in a model of antiglomerular basement membrane glomerulonephritis [104].
We have recently identified two immunodominant epitopes in the neutral endopeptidase antigen that are specifically recognized by the mothers’ antibodies [105]. Because future pregnancies in neutral-endopeptidase-immunized mothers are at high risk for the fetus [106], epitope-driven therapies including induction of mucosal tolerance are needed in addition to nonspecific immunosuppressive therapy based on intravenous Ig and high-dose corticosteroids. A similar approach could be used in idiopathic membranous nephropathy once the target (podocyte) antigen is identified. In patients with immunologically active glomerular disease, a combination of nonspecific and antigen/epitope-driven therapies should be envisaged. For instance, the effect of anti-CD20 monoclonal antibodies on Ig production could be completed by peptide-based immunotherapy aimed at reducing specifically the synthesis of antipodocyte antibody.
In conclusion, substantial progresses have been made in the past few years in understanding the pathophysiology of human membranous nephropathy. The first human podocyte antigen has been identified. Antineutral endopeptidase antibodies do not cause idiopathic membranous nephropathy, but the experimental and human data strongly suggest that most antigenic targets sit at the podocyte membrane, where they should be searched for. Translational research in this area should soon lead to assays of circulating pathogenic antibodies and to better targeted therapies aimed at decreasing specifically their production.
References
Wasserstein AG (1997) Membranous glomerulonephritis. J Am Soc Nephrol 8:664–674
Glassock RJ (2003) Diagnosis and natural course of membranous nephropathy. Semin Nephrol 23:324–332
Glassock RJ (2004) The treatment of idiopathic membranous nephropathy: a dilemma or a conundrum? Am J Kidney Dis 44:562–566
Perna A, Schieppati A, Zamora J, Giuliano GA et al (2004) Immunosuppressive treatment for idiopathic membranous nephropathy: a systematic review. Am J Kidney Dis 44:385–401
Ruggenenti P, Chiurchiu C, Brusegan V, Abbate M, Perna A, Filippi C, Remuzzi G (2003) Rituximab in idiopathic membranous nephropathy: a one-year prospective study. J Am Soc Nephrol 14:1851–1857
Cunningham PN, Quigg RJ (2005) Contrasting roles of complement activation and its regulation in membranous nephropathy. J Am Soc Nephrol 16:1214–1222
Imai H, Hamai K, Komatsuda A, Ohtani H, Miura AB (1997) IgG subclasses in patients with membranoproliferative glomerulonephritis, membranous nephropathy, and lupus nephritis. Kidney Int 51:270–276
Noel LH, Aucouturier P, Monteiro RC, Preud’Homme JL, Lesavre P (1988) Glomerular and serum immunoglobulin G subclasses in membranous nephropathy and anti-glomerular basement membrane nephritis. Clin Immunol Immunopathol 46:186–194
Heymann W, Hackel DB, Harwood S, Wilson SGF, Hunter JL (1959) Production of nephrotic syndrome in rats by Freund’s adjuvants and rat kidney suspension. Proc Soc Exp Biol Med 100:660–664
Van Damme BJ, Fleuren GJ, Bakker WW, Vernier RL, Hoedemaeker PJ (1978) Experimental glomerulonephritis in the rat induced by antibodies directed against tubular antigens. V. Fixed glomerular antigens in the pathogenesis of heterologous immune complex glomerulonephritis. Lab Invest 38:502–510
Couser WG, Steinmuller DR, Stilmant MM, Salant DJ, Lowenstein LM (1978) Experimental glomerulonephritis in the isolated perfused rat kidney. J Clin Invest 62:1275–1287
Kerjaschki D, Farquhar MG (1982) The pathogenic antigen of Heymann nephritis is a membrane glycoprotein of the renal proximal tubule brush border. Proc Natl Acad Sci U S A 79:5557–5561
Kerjaschki D, Farquhar MG (1983) Immunocytochemical localization of the Heymann nephritis antigen (gp330) in glomerular epithelial cells of normal Lewis rats. J Exp Med 157:667–686
Allegri L, Brianti E, Chatelet F, Manara GC, Ronco P, Verroust P (1986) Polyvalent antigen-antibody interactions are required for the formation of electron-dense immune deposits in passive Heymann’s nephritis. Am J Pathol 125:1–6
Christensen EI, Birn H (2002) Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol 3:256–266
Makker SP, Tramontano A (2006) Differential capacity of anti-RAP and anti-megalin antibodies to produce progressive passive Heymann nephritis-implications for the pathogenesis of idiopathic human membranous glomerulonephritis. J Pathol 210:282–287
Saito A, Pietromonaco S, Loo AKC, Farquhar MG (1994) Complete cloning and sequencing of rat gp330/“megalin,” a distinctive member of the low density lipoprotein receptor family. Proc Natl Acad Sci U S A 91:9725–9729
Raychowdhury R, Zheng G, Brown D, McCluskey RT (1996) Induction of Heymann nephritis with a gp330/megalin fusion protein. Am J Pathol 148:1613–1623
Saito A, Yamazaki H, Rader K, Nakatani A, Ullrich R, Kerjaschki D, Orlando RA, Farquhar MG (1996) Mapping rat megalin: the second cluster of ligand binding repeats contains a 46-amino acid pathogenic epitope involved in the formation of immune deposits in Heymann nephritis. Proc Natl Acad Sci U S A 93:8601–8605
Yamazaki H, Ullrich R, Exner M, Saito A, Orlando RA, Kerjaschki D, Farquhar MG (1998) All four putative ligand-binding domains in megalin contain pathogenic epitopes capable of inducing passive Heymann nephritis. J Am Soc Nephrol 9:1638–1644
Oleinikov AV, Feliz BJ, Makker SP (2000) A small N-terminal 60-kD fragment of gp600 (megalin), the major autoantigen of active Heymann nephritis, can induce a full-blown disease. J Am Soc Nephrol 11:57–64
Tramontano A, Knight T, Vizzuso D, Makker SP (2006) Nested N-terminal megalin fragments induce high-titer autoantibody and attenuated Heymann nephritis. J Am Soc Nephrol 17:1979–1985
Tramontano A, Makker SP (2004) Conformation and glycosylation of a megalin fragment correlate with nephritogenicity in Heymann nephritis. J Immunol 172:2367–2373
Shah P, Tramontano A, Makker SP (2007) Intramolecular spreading in Heymann nephritis. J Am Soc Nephrol (in press)
Ronco P, Allegri L, Melcion C, Pirotsky E, Appay MD, Bariety J, Pontillon F, Verroust P (1984) A monoclonal antibody to brush border and passive Heymann nephritis. Clin Exp Immunol 55:319–332
Ronco P, Allegri L, Brianti E, Chatelet F, Van Leer EHG, Verroust P (1989) Antigenic targets in epimembranous glomerulonephritis. Experimental data and potential application in human pathology. Appl Pathol 7:85–98
Assmann KJ, van Son JP, Dijkman HB, Koene RA (1992) A nephritogenic rat monoclonal antibody to mouse aminopeptidase A. Induction of massive albuminuria after a single intravenous injection. J Exp Med 175:623–635
Chatelet F, Brianti E, Ronco P, Roland J, Verroust P (1986) Ultrastructural localization by monoclonal antibodies of brush border antigens expressed by glomeruli. I. Renal distribution. Am J Pathol 122:500–511
Chatelet F, Brianti E, Ronco P, Roland J, Verroust P (1986) Ultrastructural localization by monoclonal antibodies of brush border antigens expressed by glomeruli. II. Extrarenal distribution. Am J Pathol 122:512–519
Assmann KJ, Tangelder MM, Lange WP, Tadema TM, Koene RA (1983) Membranous glomerulonephritis in the mouse. Kidney Int 24:303–312
Assmann KJ, Ronco P, Tangelder MM, Lange WP, Verroust P, Koene RA (1985) Comparison of antigenic targets involved in antibody-mediated membranous glomerulonephritis in the mouse and rat. Am J Pathol 121:112–122
Matsuo S, Fukatsu A, Taub ML, Caldwell PR, Brentjens JR, Andres G (1987) Glomerulonephritis induced in the rabbit by antiendothelial antibodies. J Clin Invest 79:1798–1811
Ronco P, Ardaillou N, Verroust P, Lelongt B (1994) Pathophysiology of the podocyte: A target and a major player in glomerulonephritis. Adv Nephrol Necker Hosp 23:91–131
Debiec H, Guigonis V, Mougenot B, Decobert F, Haymann JP, Bensman A, Deschenes G, Ronco P (2002) Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med 346:2053–2060
Debiec H, Nauta J, Coulet F, van der Burg M, Guigonis V, Schumans T, de Heer E, Soubrier F, Janssen F, ronco P (2004) Role of truncating mutations in MME gene in feto-maternal allo-immunization and neonatal glomerulopathies. Lancet 364:1252–1259
Nauta J, de Heer E, Baldwin WM 3rd, ten Kate FJ, v d Heijden AJ, Wolff ED (1990) Transplacental induction of membranous nephropathy in a neonate. Pediatr Nephrol 4:111–116
Tauc M, Châtelet F, Verroust P, Vandewalle A, Poujeol P, Ronco P (1988) Characterization of monoclonal antibodies specific for rabbit renal brush-border hydrolases: application to immunohistological localization. J Histochem Cytochem 36:523–532
Matsuo S, Caldwell PRB, Brentjens JR, Andres G (1985) In vivo interactions of antibodies with cell surface antigens. A mechanism responsible for in situ formation of immune deposits in the zona pellucida of rabbit oocytes. J Clin Invest 75:1369–1380
Lu B, Figini M, Emanueli C, Geppetti P, Grady EF, Gerard NP, Ansell J, Payan DG, Gerard C, Bunnett N (1997) The control of microvascular permeability and blood pressure by neutral endopeptidase. Nat Med 3:904–907
Ikeda K, Emoto N, Raharjo SB, Nurhantari Y, Saiki K, Yokoyama M, Matsuo M (1999) Molecular identification and characterization of novel membrane-bound metalloprotease, the soluble secreted form of which hydrolyzes a variety of vasoactive peptides. J Biol Chem 274:32469–32477
Bonvouloir N, Lemieux N, Crine P, Boileau G, DesGroseillers L (2001) Molecular cloning, tissue distribution, and chromosomal localization of MMEL2, a gene coding for a novel human member of the neutral endopeptidase-24.11 family. DNA Cell Biol 20:493–498
Gough J, Yilmaz A, Yilmaz S, Benediktsson H (2005) Recurrent and de novo glomerular immune-complex deposits in renal transplant biopsies. Arch Pathol Lab Med 129:231–233
Lin J et al (2001) Membranous glomerulopathy associated with graft-versus-host disease following allogeneic stem cell transplantation: report of 2 cases and review of the literature. Am J Nephrol 21:351–356
Rossi L et al (2001) Membranous glomerulonephritis after haematopoietic cell transplantation for multiple myeloma. Nephron 88:260–263
Miyazaki Y et al (2003) Membranous nephropathy associated with donor lymphocyte infusion following allogeneic bone marrow transplantation. Int J Hematol 78:262–265
Tsutsumi C et al (2004) Membranous nephropathy after allogeneic stem cell transplantation: report of 2 cases. Int J Hematol 79:193–197
Stevenson WS et al (2005) Nephrotic syndrome after stem cell transplantation. Clin Transplant 19:141–144
Ikee R et al (2004) Recurrent nephrotic syndrome associated with graft-versus-host disease. Bone Marrow Transplant 34:1005–1006
Bruijn JA et al (1988) Murine chronic graft-versus-host disease as a model for lupus nephritis. Am J Pathol 130:639–641
Bianchi DW et al (1996) Male fetal progenitor cells persist in maternal blood for as long as 27 years postpartum. Proc Natl Acad Sci U S A 93:705–708
Maloney S et al (1999) Microchimerism of maternal origin persists into adult life. J Clin Invest 104:41–47
Adams KM, Nelson JL (2004) Microchimerism: an investigative frontier in autoimmunity and transplantation. JAMA 291:1127–1131
Khosrotehrani K, Bianchi DW (2003) Fetal cell microchimerism: helpful or harmful to the parous woman? Curr Opin Obstet Gynecol 15:195–199
Nagahama K et al (2005) Possible role of autoantibodies against nephrin in an experimental model of chronic graft-versus-host disease. Clin Exp Immunol 141:215–222
Ronco PM (1999) Paraneoplastic glomerulopathies: New insights into an old entity (clinical conference). Kidney Int 56:355–377
Hörl WH, Kerjaschki D (2000) Membranous glomerulonephritis (MGN). J Nephrol 13:291–316
Winfield JB, Faiferman I, Koffler D (1977) Avidity of anti-DNA antibodies in serum and IgG glomerular eluates from patients with systemic lupus erythematosus. Association of high avidity antinative DNA antibody with glomerulonephritis. J Clin Invest 59:90–96
van Bruggen MC, Kramers C, Walgreen B, Elema JD, Kallenberg CG, van den Born J, Smeenk RJ, Assmann KJ, Muller S, Monestier M, Berden JH (1997) Nucleosomes and histones are present in glomerular deposits in human lupus nephritis. Nephrol Dial Transplant 12:57–66
Jordan SC, Buckingham B, Sakai R, Olson D (1991) Studies of immune-complex glomerulonephritis mediated by human thyroglobulin. N Engl J Med 304:1212–1215
Takekoshi Y, Tanaka M, Miyakawa Y, Yoshizawa H, Takahashi K, Mayumi M (1979) Free “small” and IgG-associated “large” hepatitis B e antigen in the serum and glomerular capillary walls of two patients with membranous glomerulonephritis. N Engl J Med 300:814–819
Lai KN, Li PK, Lui SF, Au TC, Tam JS, Tong KL, Lai FM (1991) Membranous nephropathy related to hepatitis B virus in adults. N Engl J Med 324:1457–1463
Gamble CN, Reardan JB (1975) Immunopathogenesis of syphilitic glomerulonephritis. Elution of antitreponemal antibody from glomerular immune-complex deposits. N Engl J Med 292:449–454
Nagashima R, Maeda K, Yuda F, Kudo K, Saitoh M, Takahashi T (1997) Helicobacter pylori antigen in the glomeruli of patients with membranous nephropathy. Virchows Arch 431:235–239
Chen JS, Chen A, Chang LC, Chang WS, Lee HS, Lin SH, Lin YF (2004) Mouse model of membranous nephropathy induced by cationic bovine serum albumin: antigen dose-response relations and strain differences. Nephrol Dial Transplant 19:2721–2728
Naruse T, Kitamura D, Miyakawa Y, Shibata S (1973) Deposition of renal tubular epithelial antigens along the renal glomerular capillary walls of patients with membranous glomerulonephritis. J Immunol 110:1163–1169
Douglas MFS, Rabideau DP, Schwartz MM, Lewis EJ (1981) Evidence of autologous immune complex nephritis. N Engl J Med 305:1326–1329
Zanetti M, Mandet C, Duboust A, Bedrossian J, Bariety J (1981) Demonstration of a passive Heymann-nephritis like mechanism in human kidney transplants. Clin Nephrol 15:272–288
Ivanyi B, Haszon I, Endreffy E, Szenohradszky P, Petri IB, Kalmar T, Butkowski RJ, Charonis AS, Turi S (1998) Childhood membranous nephropathy, circulating antibodies to the 58-kD TIN antigen, and anti-tubular basement membrane nephritis: an 11-year follow-up. Am J Kidney Dis 32:1068–1074
Nelson TR, Charonis AS, McIvor RS, Butkowski RJ (1995) Identification of a cDNA encoding tubulointerstitial nephritis antigen. J Biol Chem 270:16265–16270
Butkowski RJ, Kleppel MM, Katz A, Michael AF, Fish AJ (1991) Distribution of tubulointerstitial nephritis antigen and evidence for multiple forms. Kidney Int 40:838–846
Habib R, Beziau A, Goulet O, Blanche S, Niaudet P (1993) Renal involvement in autoimmune enteropathies. Ann Pediatr 40:103–107
Couser WG, Salant DJ (1980) In situ immune complex formation and glomerular injury. Kidney Int 17:1–13
Salant DJ, Belok S, Madaio MP, Couser WG (1980) A new role for complement in experimental membranous nephropathy in rats. J Clin Invest 66:1339–1350
Baker PJ, Ochi RF, Schulze M, Johnson RJ, Campbell C, Couser WG (1989) Depletion of C6 prevents development of proteinuria in experimental membranous nephropathy in rats. Am J Pathol 135:185–194
Cybulsky AV, Quigg RJ, Salant DJ (1986) The membrane attack complex in complement-mediated glomerular epithelial cell injury: formation and stability of C5b-9 and C5b-7 in rat membranous nephropathy. J Immunol 137:1511–1516
Ronco P, Debiec H (2005) Molecular pathomechanisms of membranous nephropathy: from Heymann nephritis to alloimmunization. J Am Soc Nephrol 16:1205–1213
Noble B, Van Liew JB, Andres GA, Brentjens JR (1984) Factors influencing susceptibility of LEW rats to Heymann nephritis. Clin Immunol Immunopathol 30:241–254
Doi T, Mayumi M, Kanatsu K, Suehiro F, Hamashima Y (1984) Distribution of IgG subclasses in membranous nephropathy. Clin Exp Immunol 58:57–62
Haas M (1994) IgG subclass deposits in glomeruli of lupus and nonlupus membranous nephropathies. Am J Kidney Dis 23:358–364
Doi T, Kanatsu K, Nagai H, Suehiro F, Kuwahara T, Hamashima Y (1984) Demonstration of C3d deposits in membranous nephropathy. Nephron 37:232–235
Moll S, Miot S, Sadallah S, Gudat F, Mihatsch MJ, Schifferli JA (2001) No complement receptor 1 stumps on podocytes in human glomerulopathies. Kidney Int 59:160–168
Neale TJ, Ullrich R, Ojha P, Poczewski H, Verhoeven AJ, Kerjaschki D (1993) Reactive oxygen species and neutrophil respiratory burst cytochrome b558 are produced by kidney glomerular cells in passive Heymann nephritis. Proc Natl Acad Sci U S A 90:3645–3649
Neale TJ, Ojha PP, Exner M, Poczewski H, Ruger B, Witztum JL, Davis P, Kerjaschki D (1994) Proteinuria in passive Heymann nephritis is associated with lipid peroxidation and formation of adducts on type IV collagen. J Clin Invest 94:1577–1584
Kerjaschki D (2004) Pathomechanisms and molecular basis of membranous glomerulopathy. Lancet 364:1194–1196
Urushihara M, Kagami S, Kuhara T, Tamaki T, Kuroda Y (2002) Glomerular distribution and gelatinolytic activity of matrix metalloproteinases in human glomerulonephritis. Nephrol Dial Transplant 17:1189–1196
McMillan JI, Riordan JW, Couser WG, Pollock AS, Lovett DH (1996) Characterization of a glomerular epithelial cell metalloproteinase as matrix metalloproteinase-9 with enhanced expression in a model of membranous nephropathy. J Clin Invest 97:1094–1101
Saran AM, Yuan H, Takeuchi E, McLaughlin M, Salant DJ (2003) Complement mediates nephrin redistribution and actin dissociation in experimental membranous nephropathy. Kidney Int 64:2072–2078
Doublier S, Ruotsalainen V, Salvidio G, Lupia E, Biancone L, Conaldi PG, Reponen P, Tryggvason K, Camussi G (2001) Nephrin redistribution on podocytes is a potential mechanism for proteinuria in patients with primary acquired nephrotic syndrome. Am J Pathol 158:1723–1731
Ronco P (2007) Proteinuria: is it all in the foot? J Clin Invest 117:2079–2082
Topham PS, Haydar SA, Kuphal R, Lightfoot JD, Salant DJ (1999) Complement-mediated injury reversibly disrupts glomerular epithelial cell actin microfilaments and focal adhesions. Kidney Int 55:1763–1775
Yuan H, Takeuchi E, Taylor GA, McLaughlin M, Brown D, Salant DJ (2002) Nephrin dissociates from actin, and its expression is reduced in early experimental membranous nephropathy. J Am Soc Nephrol 13:946–956
Sever S et al (2007) Proteolytic processing of dynamin by cytoplasmic cathepsin L defines a mechanism for proteinuric kidney disease. J Clin Invest 117:2095–2104
Takano T, Cybulsky AV (2000) Complement C5b-9-mediated arachidonic acid metabolism in glomerular epithelial cells: role of cyclooxygenase-1 and -2. Am J Pathol 156:2091–2101
Torbohm I, Schonermark M, Wingen AM, Berger B, Rother K, Hansch GM (1990) C5b-8 and C5b-9 modulate the collagen release of human glomerular epithelial cells. Kidney Int 37:1098–1104
Kuroki A, Shibata T, Honda H, Totsuka D, Kobayashi K, Sugisaki T (2002) Glomerular and serum IgG subclasses in diffuse proliferative lupus nephritis, membranous lupus nephritis, and idiopathic membranous nephropathy. Intern Med 41:936–942
Ohtani H, Wakui H, Komatsuda A, Okuyama S, Masai R, Maki N, Kigawa A, Sawada K, Imai H (2004) Distribution of glomerular IgG subclass deposits in malignancy-associated membranous nephropathy. Nephrol Dial Transplant 19:574–579
Simister NE, Story CM (1997) Human placental Fc receptors and the transmission of antibodies from mother to fetus. J Reprod Immunol 37:1–23
Clark MR (1997) IgG effect or mechanisms. Chem Immunol 65:88–110
Lucisano YM, Lachmann PJ (1991) The effect of antibody isotype and antigenic epitope density on the complement-fixing activity of immune-complexes: a systematic study using chimeric anti-NIP antibodies with human Fc regions. Clin Exp Immunol 84:1–8
Kerjaschki D, Exner M, Ullrich R, Susani M, Curtiss LK, Witztum JL, Farquhar MG, Orlando RA (1997) Pathogenic antibodies inhibit the binding of apolipoproteins to megalin/gp330 in passive Heymann nephritis. J Clin Invest 100:2303–2309
Dussaule JC, Stefanski A, Bea ML, Ronco P, Ardaillou R (1993) Characterization of neutral endopeptidase in vascular smooth muscle cells of rabbit renal cortex. Am J Physiol 264:F45–F52
Sumimoto M, Shen R, Nanus DM (2005) Involvement of neutral endopeptidase in neoplastic progression. Biochim Biophys Acta 1751:52–59
Terawaki SI, Kitano K, Hakoshima T (2007) Structural basis for type II membrane protein binding by ERM proteins revealed by the radixin-neutral endopeptidase 24–11 (NEP) complex. J Biol Chem 282:19854–19862
Reynolds J, Prodromidi EI, Juggapah JK, Abbott DS, Holthaus KA, Kalluri R, Pusey CD (2005) Nasal administration of recombinant rat alpha3(IV)NC1 prevents the development of experimental autoimmune glomerulonephritis in the WKY rat. J Am Soc Nephrol 16:1350–1359
Debiec H, Luimula P, Lefeu F, Nortier JL, Ronco P (2006) Identification of B-cell epitopes on neutral endopeptidase in feto-maternal allo-immunization with antenatal glomerulopathies. J Am Soc Nephrol 17:F–PO065 (abstract)
Nortier JL, Debiec H, Tournay Y, Mougenot B, Noël JC, Deschodt-Lanckman MM, Janssen F, Ronco P (2005) Neonatal Disease in neutral endopeptidase alloimmunization: lessons for pregnancy management and Immunological monitoring. Pediatric Nephrol 21:1399–1405
Acknowledgment
The research of the authors is funded by grants from GIS-Institut des Maladies Rares, Agence de la Biomédecine, Programme Hospitalier de Recherche Clinique, INSERM, Fondation pour la Recherche Médicale, AURA (Association pour l’Utilisation du Rein Artificiel), AMGEN France, and Genzyme Renal Innovations Program (GRIP). We thank Christine Vial for assistance in editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ronco, P., Debiec, H. Target antigens and nephritogenic antibodies in membranous nephropathy: of rats and men. Semin Immunopathol 29, 445–458 (2007). https://doi.org/10.1007/s00281-007-0091-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-007-0091-2