
Abstract
Mannan-binding lectin (MBL)-associated serine proteases (MASPs) circulate in plasma as zymogens in complexes with MBL and with L- and H-ficolin. Upon binding of MBL or ficolin to pathogen-associated molecular patterns, the MASPs are activated. MASP-2 can now cleave C4 and C2 to generate the C3 convertase, C4bC2b. The functions of the other two MASPs, MASP-1 and MASP-3 have not been elucidated. MASP-1 can cleave C2, and with low efficiency also C3, and may serve a function through direct C3 activation. No natural substrate for MASP-3 has been identified. MBL deficiency, occurring at a frequency of about 10%, is the most common congenital immunodeficiency and is associated with susceptibility to infections and autoimmune disorders. Inherited MASP-2 deficiency has been described as the result of a mutation causing the exchange of aspartic acid with a glycine at position 105, a position in the first domain, CUB1, involved in calcium binding. This mutation abolishes the binding to MBL and ficolins, and deprives MASP-2 of functional activity. The index case suffered from recurrent severe infections and autoimmune reactions. The gene frequency of the mutation among Caucasians is 3.6%. It is not found in Chinese, who present a different mutation also associated with MASP-2 deficiency.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
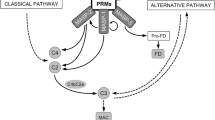
The complement system provides an important effector mechanism of innate humoral defence. Activation of the complement system proceeds through three different pathways converging in the activation of C3. The classical pathway is typically initiated after antigen recognition by antibodies, the alternative pathway relies on interference by foreign substances in a delicate activation–inhibition balance and the third pathway, the mannan-binding lectin (MBL) or lectin pathway, is initiated when one of the molecules MBL, L-ficolin or H-ficolin recognises ligands arranged in patterns characteristic of microbial surfaces, pathogen-associated molecular patterns or PAMPs [1–4] (Fig. 1). In circulation, MBL and the ficolins are found in complexes with four structurally related proteins, the MBL-associated serine proteases (MASPs) 1, 2 and 3, and MAp19, which is a truncated version of MASP-2. The four MBL-associated proteins are generated from only two genes, the MASP-1/3 gene encodes MASP-1 and MASP-3 [5], whereas the MASP-2/MAp19 gene gives rise to MASP-2 and MAp19 [6]. In both instances, alternative splicing is responsible for the generation of two different mRNAs from a single primary transcript. MASP-2 sequentially cleaves C4 and C2 upon binding of MBL/MASP-2 complexes to microbial surfaces [7], whereas the functions of MASP-1, MASP-3 and MAp19 remain to be established. The frequency of genetically determined MBL deficiency is significantly increased in patients presenting a variety of infections or autoimmune disorders, indicating the importance of the MBL pathway in the first line of defence. Recently, a patient presenting repeated infections and autoimmune manifestations was found to lack MASP-2 function, suggesting that MASP-2 deficiency represents a new congenital immunodeficiency [8]. Because MASP-2 is involved in the biological activity of both MBL and the ficolins, one might suspect MASP-2 deficiency to have broader consequences than MBL deficiency. Below, we shall describe briefly the pattern recognition molecules (PRMs), MBL, L-ficolin and H-ficolin before moving on to the MASPs.

Overview of the complement system with focus on the MBL pathway. MBL/MASP-2 complexes generate the C3 convertase, C4bC2b. MBL/MASP-1 complexes may directly activate C3. It is not known to what extent components of the alternative pathway are involved in this process. A novel aspect is that complexes of ficolin and MASP also appear to be able to activate complement
Mannan-binding lectin
MBL is a plasma protein of hepatic origin [9, 10] and belongs to a family of proteins known as the collectins. Collectins are oligomers constructed of polypeptide chains characterised by a carbohydrate recognition domain (CRD) attached to a collagen-like region. Three identical polypeptide chains assemble into subunits, which associate into higher oligomeric forms [1]. Human MBL is encoded by the MBL2 gene [11], whereas MBL1 is a pseudo gene [12]. Transcription, splicing and translation of the MBL2 gene generate a polypeptide chain of an estimated weight of 24 kDa. Due to post-translational modifications, the polypeptide runs with an apparent molecular weight of 32 kDa on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) [13, 14]. The structural subunit consists of three polypeptides joined N-terminally by disulfide linkage, followed by a helical collagen-like region and a cluster of three C-terminal CRDs [15, 16]. The subunits associate via disulfide bonding in the N-terminal region into higher oligomers ranging from two to more than six subunits. On gel permeation chromatography, MBL elutes in a fairly symmetrical peak covering the range of about 400 to 600 kDa, and a similar impression is gathered from sucrose gradient density centrifugation. We have previously termed the dominant (on SDS-PAGE) oligomeric forms in serum, MBL-I, MBL-II, MBL-III and MBL-IV (in order of increasing size) [5]. The main C4 activating activity, i.e. complex formation with MASP-2, appears to be associated with MBL-II [5], now known to represent the tetramer [17]. The molecular weights were by mass spectrometry found to be 228 kDa for MBL-I and 305 kDa for MBL-II [17]. We have observed similar values by low angle scattering, where MBL-I and MBL-II were estimated to be 220 and 300 kDa, respectively (unpublished data). Examined by electron microscopy or atomic force microscopy, the oligomers show sertiform structures [1, 18].
A commonly used term for MBL, mannose-binding lectin, indicates a special selectivity for mannose. However, MBL is not specific for this particular sugar [19]. The primary determinant for the distinct carbohydrate selectivity of MBL is the presence of horizontal hydroxyl groups at the 3′ and 4′ position of the pyranose ring (as in, e.g. d-mannose, N-acetyl-d-glucosamine, d-glucose and l-fucose), whereas it does not bind to d-galactose with a vertical hydroxyl group at the 4′ position [1, 14, 20]. The hexoses must be presented at terminal non-reducing positions and must be clustered in a pattern to obtain high avidity interactions critical for the biological function of MBL. The dissociation constant for the interaction between one CRD and a hexose is weak, in the order of 10 mM, whereas MBL binds with high avidity to polyvalent carbohydrates such as mannan (K d∼2 nM) [10, 21].
The concentration of MBL in plasma ranges 1000-fold from 5 ng to more than 5 μg per milliliter plasma and is not normally distributed [22]. The median concentration is about 1.5 μg/ml. The inter-individual differences are to a large extent explained by polymorphisms in the promotor region and in exon 1 of the MBL2 gene [23–27]. However, the constitutional MBL concentration may vary tenfold between individuals with identical genotypes [28, 29], and there is a minor (two to threefold), sluggish acute phase response [30]. In their seminal paper, Super et al. [31] reported low MBL levels in children with opsonising deficiency and repeated, serious infections. This observation has since been supported by numerous investigations [32, 33]. Low MBL levels have also been reported to be associated with a variety of autoimmune disorders [32, 34, 35]. The high incidence of low MBL levels (one in four Caucasians have less than 0.5 μg/ml) indicates that MBL insufficiency is only manifest under certain conditions when also other elements of the immune defence function suboptimally [36–38]. The high prevalence of deficiency naturally leads to speculations on MBL deficiency being advantageous in certain infections or other diseases, but so far only few and conflicting data are available.
Ficolins
The ficolins are a group of proteins, which, like MBL, bind to PAMPs. Three ficolins are found in humans: L- and M-ficolin, which are more than 80% identical, and H-ficolin [39]. There is no significant sequence similarity to MBL (apart from the collagen-like region), but the gross structures show strikingly similar sertiform shapes.
Like MBL, ficolins are composed of subunits of three identical polypeptide chains with a collagenous part and three globular ligand-recognising domains. In the ficolins, these domains are fibrinogen-like (FBG) domains [40, 41].
L- and H-ficolin are humoral factors synthesised by hepatocytes and secreted into the blood circulation (H-ficolin has also been observed in bronchial/alveolar fluid and in bile) [41–43], whereas M-ficolin is reportedly confined to the surface of peripheral blood monocytes [44, 45] (see note added in proof).
Ficolins are suspected to play a part in the innate immune defence. This supposition is largely based on analogy with MBL: structure, binding to microorganisms and association with MASPs, thus the capacity of activating the complement system [46–48]. The ficolins are not lectins in the standard definition of this term, i.e. proteins specifically binding carbohydrates. Thus H- and L-ficolin binds indiscriminately to acetyl groups [49 and unpublished results]. It may seem somewhat confusing to use the term ‘lectin pathway’ as a common denominator for complement activation through these proteins.
Congenital ficolin deficiencies have not been described [45]. Inaba et al. [50] quantified H-ficolin in 170,000 persons and found deficiency in four systemic lupus erythematosus (SLE) patients, probably secondary to the development of anti-H-ficolin auto-antibodies. One paper reports on lower levels of L-ficolin being associated with recurrent respiratory infections in children [51].
MBL-associated serine proteases
MBL and the ficolins exert biological activity by themselves through aggregation of targets and possibly through specific receptors on phagocytic cells capable of recognising MBL or ficolin coated (opsonised) targets [41, 52, 53]. However, it appears that the main function is via activation of complement with the ensuing direct killing or opsonisation of the target and the attraction of cells of the immune system. The seminal observation of complement activation by MBL was made in 1987 by Ikeda et al. [54]. Matsushita and Fujita [55] demonstrated the presence of a new complement enzyme, termed MBL-associated serine protease, which was thought to mediate the formation of the C3 convertase, C4bC2b. This activity was shown by Thiel et al. [56] to be mediated by another MASP, termed MASP-2 (the one described by Matsushita and Fujita is now termed MASP-1). Later, a third MASP, MASP-3 [5], was identified as well as a small, separately synthesised fragment of MASP-2, termed MAp19 or sMAP [6, 57]
Initially, it was thought that the activation of the MASP-1 and MASP-2 zymogens in complex with MBL was equivalent to the sequential activation of C1r and C1s within the C1 complex of the classical pathway. However, experiments conducted with recombinant MASP-2 showed that complexes of MBL and MASP-2 alone were sufficient for the generation of the C4bC2b convertase [58].
Although all four proteins were initially detected in complex with MBL, they also associate with L- and H-ficolin [47, 48, 59]. The interaction of several proteins with various recognition molecules adds to the complexity of the MBL-ficolin pathway as compared to the classical activation pathway, which is triggered only by the C1 complex. The stoichiometry of the C1 complex and the functional properties of its individual serine proteases is well defined, whereas the stoichiometric composition of the MBL/MASP and ficolin/MASP complexes remains unresolved. Despite functional and structural differences between the initiation complexes of the MBL and the classical pathway, the serine proteases share many common features.
The evolutionary aspects of MASPs have recently been discussed in detail [60, 61]. In brief, cDNA sequences of MASPs have been cloned in various species of vertebrates, e.g. in lamprey, shark, carp, chicken, mouse and humans, and from two invertebrates (amphioxus and ascidian) as well. Recently, it was found that chickens have MASP-2 and MAp19 with a gene organisation identical to those of human and mouse [62]. MASP-3 in chickens is very similar (75% identity) to mammalian MASP-3, but in the chicken, the six exons encoding the MASP-1 serine protease domain are absent, and birds thus have only MASP-3, MASP-2 and MAp19.
MBL-associated proteins: genetic organisation and sites of synthesis
Alternative splicing of the primary transcript of a single gene (MASP1/3) on chromosome 3q27–q28 yields mRNA for MASP-1 and MASP-3 [5] (Fig. 2). The gene encompasses 17 exons, of which the first 10 encode the non-catalytic N-terminal domains of both the MASP-1 and MASP-3 polypeptide (the A chain). The linker region and the C-terminal serine protease domain of MASP-1 are encoded by six separate exons, whereas for MASP-3, this is encoded by a single exon [5, 63, 64]. Northern blot analysis revealed the liver as the primary site of MASP-1 and MASP-3 synthesis [65]. It was later confirmed that in humans, rats and mice, MASP-1 expression is liver specific, whereas MASP-3 is expressed in the liver and in non-hepatic tissues, including the spleen, lung, small intestine, thymus and brain [62]. MASP-2 and MAp19 are also the products of mRNAs generated by alternative splicing of a single primary RNA transcript [6]. The human MASP-2/MAp19 gene has been mapped to chromosome 1p36.2–3 and encompasses 12 exons [66]. MASP-2 mRNA is encoded by 11 of these exons, of which a single one encodes the linker region and the serine protease domain. Exon 5 of the MASP-2/MAp19 gene allows for generation of the MAp19 mRNA splice variant encompassing code for the first two common domains and four additional C-terminal amino acids characteristic of MAp19 [66]. The liver is the only tissue in which MASP-2 and MAp19 mRNAs have been detected [65–67].

Genomic organization and protein structure of MASPs and MAp19. Top, the exon–intron structure of the MASP1/3 gene. A single exon encodes the MASP-3 B-chain, whereas this region in MASP-1 is encoded by six exons. The A chain of MASP-1 and MASP-3 is encoded by 10 shared exons. The asterisk denotes potential glycosylation sites. The scissile Arg–Ile bond, the disulphide linkage and the names of the domains are indicated. Bottom, the exon–intron structure of the MASP2/MAp19 gene. The MASP-2 B chain is encoded by a single exon, whereas the A chain is encoded by 10 exons. MAp19 is encoded by 5 exons. Four of these exons are shared with MASP-2, whereas exon 5 encodes four amino acids specific for MAp19 [65]
Several cell lines have been screened for MASP synthesis, and hepatocyte-like cell lines were found to express MASPs, but also cell lines representing glioma cells were producing mRNA for MASPs, suggesting that human astrocytes may be a source of MASP in the brain [68].
The structure of the MBL-associated proteins
The mature polypeptide chains of human MASP-1 and MASP-3 are composed of 699 and 728 amino acid residues, respectively, including the leader peptide of 19 amino acids. MAp19 and MASP-2 are polypeptides of 185 and 686 amino acid residues, respectively, including the 15 amino acid leader sequence [65]. The MASP-1, MASP-2 and MASP-3 proenzymes have calculated molecular weights of 76,976, 74,153 and 81,873 Da, respectively. By Western blot analysis, the observed molecular masses are 90, 74 and 94 kDa [65], respectively, suggesting post-translational modification of MASP-1 and MASP-3. Indeed, the MASP-1 sequence contains four N-glycosylation sites, the MASP-3 seven and MASP-2 none (Fig. 2). The calculated and observed molecular mass of the MAp19 polypeptide is 19 kDa [6].
The MASPs are composed of six modules and a linker region: an N-terminal CUB domain (CUB1) followed by an epidermal growth factor (EGF)-like domain of the Ca2+-binding type, a second CUB domain (CUB2), two contiguous complement control protein modules (CCP1 and CCP2), a short linker and finally a chymotrypsin-like serine protease (SP) domain (Fig. 2). MAp19 only encompasses the CUB1, the EGF-like domain (both shared by MASP-2) and the four MAp19 specific amino acids (EQSL).
The CUB domain of about 110 amino acid residues is found in many diverse proteins [69]. The general three-dimensional structure shows two five-stranded β-parallel sheets stabilised by two disulphide bridges, resembling an elongated ellipsoid [70]. Amino acid sequence alignment reveals that the CUB1 module in human MASPs (as well as in C1r and C1s) lack the first two parallel β-strands present in the previously determined CUB structures and also the first of the two disulfide bridges [71]. It was indeed found that the MAp19 and the rat CUB1 module are made of two four-stranded anti-parallel β-sheets [71, 72] (Fig. 3). The crystal structure of the N-terminal three domains of rat MASP-2 revealed that the second CUB domain (CUB2) contained one of the beta strands missing in CUB1 (β2), and the structure is stabilised by two disulfide bridges [72].

Top, homodimeric structure of human MAp19. Top view of the structure. The two MAp19 molecules are in red and green, respectively. Ca2+ ions are represented as golden spheres. The side chains of residues involved in the intermonomer interface are indicated. Ct indicates the C-terminal end [71]. Center, structure of the activated MASP-2 CCP2-SP fragment. Residues of the catalytic triad (a.s.) are shown as sticks. The figure was kindly provided by Harmat et al. [79]. Bottom, a hypothetical model of the complexes between MAp19 and tetrameric MBL. The CUB and EGF modules of MAp19 are blue and green, respectively. The CRD domains are in red (every subunit has three CRDs, but not all are easily seen), and the collagen-like segment containing the MAp19 binding site is in grey. The structure of the MBL collagen-like segment encompassing residues 73–87 is unknown and is shown as grey dots. A bottom view of the complexes is shown [17]
Previously, it was found that CUB1–EGF of human C1s contained a novel Ca2+-binding site at the distal end of the CUB1 module comprising six oxygen ligands (two water molecules and four amino acid residues) coordinating the Ca2+ ion, and together with three additional amino acid residues, these elements make up a network of stabilising hydrogen bonds [73]. The Ca2+ ion thus appears to be an essential component for the stability of the distal part the C1s CUB1 module. The residues involved in Ca2+ binding in the C1s CUB1 module are conserved in C1r as well as in the MASPs. An essential Ca2+ ion is also associated with the distal part of the CUB1 module of MAp19 [71] and thus also of MASP-2. The functional significance of CUB1 Ca2+ binding was demonstrated by mutating amino acid residues providing Ca2+ ligands in MAp19. The mutants only showed a weak association or failed to bind to MBL and L-ficolin as measured by surface plasmon resonance spectrometry (SPR) [71]. As discussed below, a naturally occurring polymorphism, which results in loss of calcium binding, also results in loss of binding of MAp19 and MASP-2 to MBL.
The EGF domain is found in many proteins and is known to mediate protein–protein interactions. Although they often show relatively little sequence identity with EGF itself, the domains share a characteristic configuration around three disulfide bonds between six conserved cysteine residues [74]. The EGF-like domain of approximately 50 amino acid residues is folded into an elongated structure with anti-parallel double-stranded β-sheets connected by five loops [73, 75] A subset of EGF domains, including those of MASPs and C1r and C1s, bind Ca2+ by means of a five amino acid consensus sequence: Asp/Asn, Asp/Asn, Gln/Glu, Asp*/Asn*, Tyr/Phe, with the asterisk denoting post-translational β-hydroxylation [73, 76]
The CCP domain, also known as short consensus repeat (SCR), occurs widely in modular proteins, only some of which are involved in complement activation [77]. The CCP domain of approximately 60 amino acid residues is characterised by several hydrophobic amino acids and four nearly invariant cysteines and shows a common scaffold-like structure characterised by a sandwich of 6–8 β-strands, stabilised by two disulfide bridges between the conserved cysteine residues [77, 78]. The crystal structure of the second CCP (CCP2) module in conjunction with the serine protease domain of MASP-2 showed a central compact hydrophobic core enveloped by six β-strands stabilised by two disulfide bonds [79] (Fig. 3). Compared to the rigid CCP2-SP interface observed for C1s [80], MASP-2 presents increased flexibility at the CCP2-SP junction. This may partly explain that the MASP-2 dimer alone mimics the functions of C1r plus C1s in the classical pathway.
The serine protease domain
The modular enzymes of the activation complexes of complement belong to the large family of chymotrypsin-like serine proteases and show the characteristic catalytic triad of three conserved non-consecutive His/Asp/Ser residues [81] (Fig. 3). They possess an identical fold consisting of two juxtaposed six-stranded β-barrels with the three catalytic residues situated at the interface. They also show a second conserved serine residue located adjacent to the active site, which is important for the productive formation of the enzyme-substrate complex [82, 83]. The proteases are produced as zymogens in which the active site is distorted and has low catalytical efficiency. The MASPs are synthesised as single chain proenzymes. Binding of MBL/MASP or ficolin/MASP to their respective ligands activates the MASPs by promoting a proteolytic cleavage of a conserved Arg–Ile bond within the linker region, thus producing two polypeptide chains held together by disulfide linkage [65] (Fig. 2) Therefore, by reducing SDS-PAGE analysis, the MASP proenzymes and their activated counterparts migrate as one and two bands, respectively. The five N-terminal domains and the linker region prior to the cleavage site make up the A-chain, whereas the catalytic SP domain is referred to as the B-chain. The mechanism by which initial activation is accomplished remains elusive, but may be caused by mechanical stress induced by MBL or ficolin target recognition [84].
Expression of fragments of MASP-2 (i.e. CCP1-CCP2-SP, CCP2-SP) revealed auto-activation (cleavage between CCP2-SP). Efficient binding and cleavage of C2 and inhibition by C1 inhibitor could be observed for the SP domain in combination with the linker region. On the other hand, the proteolytic cleavage of C4 was, as for C1s, dependent on the presence of the CCP modules [85, 86]. Rossi et al. [87] observed auto-activation of the recombinant CCP1-CCP2-SP fragment, but not of the CCP2-SP fragment. Upon activation, the new N-terminal peptide flips over and the isoleucine residue interacts with an aspartic acid residue immediately upstream from the active site serine. This switches on the catalytic activity of the protease [88]. An aspartic acid residue is found in the catalytic cavity in all MASPs, indicating cleavage after a basic amino acid as observed for trypsin [5, 56].
The SP domain of MASP-1 differs from those of MASP-2, MASP-3 and C1r and C1s in several aspects: (1) the domain is encoded by several exons, (2) the active site serine residue is encoded by a TCN codon (N=any nucleotide) and (3) the presence of a histidine loop (a stabilising disulfide bridge between cysteine residues situated on either side of the active site histidine) as also found in chymotrypsin. In contrast, the SP domain of the other four proteases is encoded by a single exon, an AGY (Y=C or T) codon translates into the active site serine residue, and the histidine loop is absent. Whereas the intra-molecular histidine loop could have functional implications, the split exon structure and the AGY/TCN codon usage serve merely as evolutionary markers. This indicates that the AGY type lineage diverged from the TCN type before the emergence of primitive vertebrates [64]. Phylogenetic analysis suggests that a lectin-MASP (TCN type) complex in conjunction with C3 and its receptor could constitute a primitive ancestral immune system hence antedate the classical pathway of complement activation [60]. The lamprey seems to have a molecule with C1q globular head domain structure but with lectin activity and is also protease associated [89]. This may be the very beginning of the evolution into lectin and C1q-mediated complement pathways.
Dimerization of MASPs and MAp19
MASP-1, MASP-2, MASP-3 and MAp19 each form homo-dimers and individually associate with MBL and the ficolins in a Ca2+-dependent manner through the CUB1-EGF modules. The CUB1–EGF module pair in C1r and C1s is stabilised by Ca2+, and while the EGF domain of C1r binds Ca2+ with an apparent K D of only 10 mM [90], the affinity for Ca2+ of the recombinant CUB1–EGF pair shows an apparent K D of 10–16 μM [91]. Apparently, residues located outside the EGF domain either directly provide ligands for Ca2+ chelation or stabilise the Ca2+-binding site conformation.
Expression of the recombinant N-terminal two or three domains of rat MASP-1 and MASP-2 followed by analysis by ultra-centrifugation revealed the formation of stable homodimers in the presence of calcium, which were not disrupted by ethylenediaminetetraacetic acid (EDTA) [92]. The authors concluded that MASP dimer formation is Ca2+ independent in solution and involves the CUB1–EGF pair. Sedimentation velocity analysis of recombinant human MAp19 and the CUB1–EGF segments of MASP-1 and MASP-2 showed that in the presence of Ca2+ the proteins associate in homodimers [93]. The respective homodimers did not dissociate in the presence of EDTA, although the physical properties were affected as seen by a broadening sedimentation peak. Neither of the above recombinant proteins and recombinant full-length MASP-1 and MASP-2 formed heterodimers as evidenced by gel permeation chromatography and SPR [93]. The authors concluded that dimer formation was Ca2+ dependent. Recent experiments, also based on ultra-centrifugation analysis, have indicated that recombinant human MASP-3 sediments as a homodimer in the presence of Ca2+ [94]. The MASP-3 dimer was disrupted by EDTA. The crystal structure of recombinant MAp19 revealed that MAp19 forms head-to-tail dimers involving interactions between the CUB1 module of one monomer and the EGF-like domain of its counterpart [71] (Fig. 3). A Ca2+ ion in each EGF module stabilises the inter-monomer interface and is coordinated in a manner similar to that determined for CUB1–EGF of C1s [73]. Hence, a water molecule provides a link between the CUB1 and EGF modules, thereby stabilising the inter-modular interface. Taken together, the N-terminal part of MASPs mediates the formation of homodimers, but contrary to the case of C1r and C1s, no heterodimers are formed. It remains totally unexplained why apparently no heterodimers are formed between MAp19 and MASP-2 although the two molecules share the same CUB1–EGF domains. Stranger still is the case for MASP-1 and MASP-3, which are identical throughout the entire five domains of the A-chain, but still only form homodimers.
The MBL/MASP complex
Although structural similarities between MBL and the MASPs on one hand and C1q, C1r and C1s on the other hand are evident, it is useful to stress some differences: (1) the classical pathway relies on sequential activation of C1r and C1s, with C1s subsequently generating the C3 convertase, C4bC2b, by cleaving C4 and C2 [95], whereas in the MBL pathway, one molecule, MASP-2, subserves the function of C1r + C1s [58]; (2) the C1 complex is sensitive to hypertonic buffers, which, while leaving the MBL pathway intact, inhibit the binding of C1q to immune complexes and disrupt the C1 complex; (3) the stoichiometric composition of the C1 complex is well defined as one C1q in complex with one C1s-C1r-C1r-C1s tetramer, whereas a great complexity of the MBL/MASP complexes is emerging.
Fractionation of MBL/MASP complexes from normal human serum indicated preferential association of MASP-1 and MAp19 with low oligomer MBL (termed MBL-I), whereas MASP-2 and MASP-3 appeared to be associated mainly with larger MBL oligomers (MBL-II and MBL-III) [5]. MBL-I and MBL-II have now been identified as trimers and tetramers of subunits, respectively [17]. Adding to the complexity is the association of MAp19 and MASPs with L- and H-ficolin [47, 48]. It has been estimated that less than 10% of MASP-1 MASP-2 and MAp19 are in complex with MBL [4, 59, 96].
Like in the C1 complex, Ca2+ ions also play distinct roles in the MBL/MASP/MAp19 complex. A Ca2+-binding site forms an essential and well-described part of the carbohydrate-binding structure in the CRD of MBL and chelation of a Ca2+ ion by the CUB1 module of MAp19, and MASP-2 is obligate for the interaction with MBL and L-ficolin [8, 71].
Chen and Wallis [97] examined the interaction between rat MBL and rat MASP-1 and MASP-2 using an inhibition assay based on displacement of 35S-labelled CUB1–EGF–CUB2 fragments of MASP-1 and MASP-2 bound to MBL. The results showed that the affinity between MBL and full-length MASP-1 and MASP-2 was comparable to those determined for the CUB1–EGF–CUB2 fragments. The affinity of CUB1–EGF for MBL was lower than for the three module fragments. The EGF–CUB2 fragment did not bind to MBL. In accordance with this, SPR showed that the CUB1–EGF moiety of human MASP-1 and MASP-2 (and MAp19) is sufficient for Ca2+ dependent binding to immobilised MBL and L-ficolin [93, 98]. Full-length MASP-1, its CUB1–EGF–CUB2 and CUB1–EGF fragments displayed similar k on values for the interaction with MBL and L-ficolin, but the CUB1–EGF fragment showed a larger k off value resulting in an increased K D. Similar results were obtained for MASP-2, its N-terminal fragments and MAp19.
Using SPR, Teillet et al. [17] recently found that the MASPs and MAp19 interact with equal affinity with trimeric MBL (MBL-I) and tetrameric MBL (MBL-II) (K d about 2.5 nM for MASPs and 10 nM for MAp19). This contrasts curiously to the original observation of selectivity of the various MASPs and MAp19 for different MBL oligomers, and also that C4 cleaving activity was associated with MBL-II/MASP-2 complexes [5]. This observation is supported by the finding that affinity chromatography of serum on monoclonal anti-MASP-2 selectively co-purifies MBL-II (Thiel and Jensenius, unpublished data). It appears that the artificial conditions of SPR may not faithfully reproduce the complex in vivo situation.
Wallis et al. [97, 99] showed that a dimer of rat MBL-A subunits is sufficient for binding of MASPs. Recombinant MBL polypeptide chains simulating the B allotype (Gly54Asp) have lost the ability to associate with MASPs [100, 101]. The mutant polypeptides may form a subunit but are not able to form higher oligomers. Interestingly, Chen and Wallis [102] found that MBL blocks the interaction of MASP-2 with its substrate, C4, until the activation of the MBL/MASP complex through binding to carbohydrate ligand.
In summary, MASP-1, MASP-2, MASP-3 as well as MAp19 each associate Ca2+ dependently into homodimers via their CUB1–EGF module pair in a head-to-tail fashion. The MASPs and MAp19 each individually form Ca2+-dependent complexes with MBL and L-ficolin. The interaction with MBL and L-ficolin is determined by the N-terminal CUB1–EGF moiety, but is strengthened by the presence of the CUB2 module.
Substrate specificities and inhibition of the MASP
With the identification of MASP-2, the ability to cleave C4 was determined to be a feature of this protease and not MASP-1 [56]. This was concluded from experiments using minimally denaturing SDS-PAGE and Western, which potentially could affect the proteolytic activity of both MASP-1 and MASP-2. It was later supported by experiments using human recombinant MASP-2 expressed in a mammalian cell line, which showed that MASP-2 cleaves C4 and C2, generating the C3 convertase [58].
To study the proteolytic activities of MASP-1 and MASP-2 against C2, C3 and C4, both proteases were isolated from human serum by means of sequential affinity chromatography [103]. Activated MASP-1 showed proteolytic activity against C2 and C3, whereas activated MASP-2 cleaved C2 and C4. Using a different procedure Petersen et al. [104] verified a low level of proteolytic activity of MASP-1 against C3, and direct activation of C3 by MBL-I/MASP-1 has also been demonstrated [5]. Other authors using recombinant proteins found only very low C3 cleaving activity of MASP-1 and argue that this activity is likely too low to be of physiological significance [85, 105].
Employing fluorescent amide substrates, Presanis et al. [106] found MASP-1 to cleave synthetic substrates after an arginine or lysine residue, a selectivity which strongly resembles that of thrombin [85]. Although generally, cleavage specificity for synthetic oligopeptides is not evidence of natural substrate specificity, it is interesting that thrombin inhibitors (anti-thrombin III (ATIII) and an artificial peptide inhibitor) could prevent cleavage of synthetic substrate by MASP-1 [106]. MASP-1 was also found to cleave two protein substrates of thrombin, factor XIII and fibrinogen [105]. Dahl et al. [5] failed to observe proteolytic activity of natural MASP-3 against complement proteins, and it appears that recombinant MASP-3 does not undergo self-activation and that activated recombinant MASP-3 has no proteolytic activity on C2, C3 and C4 [94]. To date, no natural substrates have been found for MASP-3, but proteolytic activity was demonstrated using a synthetic substrate [94].
Several studies have addressed the control of the MBL pathway of complement. Petersen et al. [107] showed that C1 inhibitor (C1INH) could prevent C4b deposition by MBL/MASP complexes on a mannan-coated surface. Alpha-2-macroglobulin (α2M) and ATIII did not inhibit C4b deposition, although ATIII was observed to inhibit upon the addition of a physiological relevant concentration of heparin. In accordance with these results, Presanis et al. [106] recently demonstrated an inhibitory effect of C1INH and ATIII, in the presence of heparin, on the enzymatic activity of both MASP-1 and MASP-2. Matsushita et al. [103] demonstrated that C1INH forms equimolar complexes with activated human MASP-1 and MASP-2 and inhibits their proteolytic activity. This was also demonstrated using recombinant MASP-1 and MASP-2 [85, 87]. An inhibitory activity of α2M against MASP-2 is controversial. One study reported such inhibitory activity [108], whereas two other studies observed no such activity [85, 107]. However, both α2M and C1INH are associated with MBL/MASP complexes, and both are released upon activation of the complex [107]. There are also indications that α2M could be a significant inhibitor of MASP-1 activity [85, 87, 107]. MASP-3 does not react with C1INH, indicating modes of activation and control of MASP-3 different from those of MASP-1 and MASP-2 [94].
MASP and disease associations
MBL deficiency is by far the most common congenital immunodeficiency, and abundant papers have described increased frequency of MBL deficiency among patients with a history of otherwise unexplainable propensity for infections or autoimmune manifestations [32, 35]. It appeared relevant to investigate if MASP deficiency exists, and if so, what the consequences might be. We thus constructed a functional assay for the MBL pathway by estimating C4b deposition on a mannan surface [22]. Inasmuch as we include the addition of exogenous C4, this assay essentially estimates the activity of the MBL/MASP-2 complex. The assay applied on 100 normal plasmas shows strict correlation between C4b deposition and MBL concentration (p=0.98) (Fig. 4). However, when applied on samples (N=125) from suspect immuno-deficient patients, one patient was completely deficient in MBL pathway activity [8] despite having sufficient MBL (Fig. 4). The MBL/MASP complexes purified from patient plasma were devoid of both MASP-2 and MAp19 while containing MASP-1 and MASP-3 at increased levels. The MBL pathway could be reconstituted with rMASP-2, but not with rMBL. Sequence analysis of the promoter region and the exons encoding MASP-2/MAp19 revealed the patient to be homozygous for a point mutation in exon 3 of the gene, which results in the substitution of an aspartic acid residue with a glycine at position 105 in the mature MASP-2 (D105G) (D120G when including the signal peptide). This substitution, close to the C-terminal of CUB1, will affect both MASP-2 and MAp19. Western blotting indicated a reduced concentration of both proteins in serum. Wild-type and mutant-type recombinant MASP-2 were produced in parallel in a mammalian expression system to allow for examining the phenotypic consequences of the D105G substitution. The two proteins were produced at similar rates. Wild-type rMASP-2 bound to solid phase MBL and catalysed the deposition of C4b, whereas the mutant rMASP-2 did neither [8]. Identical results are seen concerning binding to L-ficolin and H-ficolin (Fig. 5).

Correlation between MBL concentration in plasma and activity of the MBL/MASP-2 complex as measured by C4b deposition on a mannan-coated surface. Despite presenting sufficient MBL levels, one suspect immuno-deficient patient was found to be deficient in MBL activity (green dot)

Binding of MASP-2 (•) and MASP-2(D105G) (○) to MBL (a), L-ficolin (b) and H-ficolin (c). Serum diluted 1/10 in a hypertonic EDTA-containing buffer was incubated in microtiter wells coated with anti-MBL, anti-L-ficolin or anti-H-ficolin antibody. This was followed by incubation with recombinant MASP-2 or recombinant MASP-2(D105G) in a Ca2+-containing buffer. Bound MASP-2 was detected with Eu3+-labelled anti-MASP-2 antibody and time resolved fluorometry
As described above, the crystal structure of the MASP-2 CUB–EGF domain has been resolved (Fig. 3), and it appears possible to explain the profound functional effect of the mutation through the observed involvement of aspartic acid residue 105 in binding of the essential Ca2+ ion (Fig. 6).

Top, the CUB1 Ca2+-binding site oxygen atoms are shown in red, and nitrogen atoms in blue. The water molecule is represented as a light blue sphere. Ionic bonds are represented by dotted lines [71]. Bottom, the nucleotide and amino acid sequences flanking the mutation in CUB1 (amino acid 105 in the mature protein)
Monoclonal anti-MASP-2 antibodies were developed and used for quantification. The patient was found to be deficient not only in MASP-2 function, but also presented a low MASP-2 concentration, about 75 ng/ml, whereas the heterozygous parents both had about 200 ng/ml. This was the case also for 11 other heterozygous individuals subsequently identified, whereas the concentration in wild-type homozygous individuals was twice this [109]. It seems likely that the inability to form complexes with MBL and the ficolins may account for the low concentration in plasma simply through decreased half life of non-complexed MASP-2, rather than being due to defects in synthesis.
Unfortunately, the index case for MASP-2 deficiency remains the only well-described case of MASP-2 deficiency. The patient is ill. Totally disabled at the age of 30 years, with several autoimmune manifestations (Table 1) and suffering from frequent infections, including pneumonia and bacteraemia. He had no symptoms until 15 years of age [8].
We have seen only one other case of MASP-2 deficiency due to homozygosity of the D105G mutation in one patient attending the lung clinic for allergic symptoms. Clearly, much more has to be done before we will know the implications of MASP-2 deficiency.
Initially, a gene frequency of 5.5% was estimated for the D105G mutation [8]. We have now extended this sample and determined 35 heterozygotes in a total of 492 Danes, a gene frequency of 3.6% and an expected frequency of homozygosity of 1 in 1,000. This is above the estimated frequency of any other known immunodeficiency, apart from MBL and IgA deficiency. The next most frequent is C2 deficiency, occurring at a rate of about 1 per 20,000 individuals [110].
We have analysed 573 Hong Kong Chinese, and none showed the D105G mutation. However, a new deficiency-associated mutation was revealed, occurring in the heterozygous state in three normal healthy persons. This Chinese mutation thus had a gene frequency of 0.26% in Hong Kong and was absent in the 492 Danes (to be published). None of the two mutations were revealed in a total of 415 South Saharan Africans. The frequency of the Hong Kong mutation is too low for conclusions to be drawn on possible occurrence in other ethnic groups. However, it seems fairly safe to conclude that the Scandinavian mutation is limited to Caucasians.
Carlsson et al. [111] studied 112 cystic fibrosis (CF) patients to evaluate the clinical significance of MBL pathway deficiencies. The MBL genes were genotyped, and the individuals were also tested for the MASP-2 D105G allele. Heterozygosity for the mutation was found in 14 patients, corresponding to an allele frequency of 6.3%. The MASP-2 level in the heterozygous CF patients (157 ng/ml) was significantly lower than in patients with wild-type genes (380 ng/ml). In 200 blood donors tested, the mutant allele frequency was 1.3% as 5 were found to be heterozygous. Again, the MASP level was lower in the heterozygous (166 ng/ml) than in the wild types (353 ng/ml). No correlation between clinical data and MASP-2 alleles was seen in the CF population.
In their investigation of the MBL pathway in patients with colorectal cancer, Ytting et al. [112] measured MASP-2 levels in serum samples taken prior to elective resection for primary colon cancer. The number and nature of post-operative infections, time to recurrence and time to death was recorded. Significantly higher MASP-2 levels were found in the 605 patients than in 150 controls (median 415 and 368 ng/ml, respectively; p=0.0002). Possibly, this might be due to an ongoing acute phase response. Contrary to the case for MBL levels [113], no significant association of MASP-2 levels with infections was seen in the cancer patient. Unexpectedly, it was found that MASP-2 levels correlated significantly with recurrence of cancer (p=0.02, trend test) and with death (p=0.0005, trend test). The median survival time of patients with MASP-2 values in the highest tertile (33%) was 29 months (95% CI 22–42), in the intermediate group it was 41 months (95% CI 32–58) and 61 months (95% CI 43–76) in the lowest tertile. The cut points were 347 and 491 ng/ml, and the medians in the three groups were 258, 416 and 646 ng/ml. Multivariate analysis showed that the MASP-2 level has independent prognostic value for survival (HR=1.5, p=0.0001). The biology behind this observation remains obscure.
We have developed an assay for MASP-3 and examined 100 samples from blood donors for MASP-3 levels without encountering any deficiencies (unpublished data). Assay for MASP-1 is only now being developed.
MASP-2 deficiency represents a new congenital immunodeficiency. Inasmuch as complement activation via H-ficolin and L-ficolin as well as via MBL will be affected, it may have more profound implications than the more frequently occurring MBL deficiency. However, further investigations are clearly required to reveal the clinical relevance of MASP-2 deficiency. Uncovering the importance of MASP-2 should also be furthered by the construction of transgenic mice lacking MAp19 and MASP-2 [114].
References
Holmskov U, Thiel S, Jensenius JC (2003) Collectins and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol 21:547
Janeway CA, Travers P, Walport M, Shlomchik M (2001) Immunobiology: the immune system in health and disease, 5th edn. Garland Publishing, New York, NY: 2001
Goldsby RA, Kindt TJ, Osborne BA (2000) Immunology, 4th edn. Freeman, New York, NY
Matsushita M, Fujita T (2001) Ficolins and the lectin complement pathway. Immunol Rev 180:78
Dahl MR, Thiel S, Matsushita M et al (2001) MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity 15:127
Stover CM, Thiel S, Thelen M et al (1999) Two constituents of the initiation complex of the mannan-binding lectin activation pathway of complement are encoded by a single structural gene. J Immunol 162:3481
Vorup-Jensen T, Jensenius JC, Thiel S (1998) MASP-2, the C3 convertase generating protease of the MBLectin complement activating pathway. Immunobiology 199:348
Stengaard-Pedersen K, Thiel S, Gadjeva M et al (2003) Inherited deficiency of mannan-binding lectin-associated serine protease 2. N Engl J Med 349:554
Kawasaki T, Etoh R, Yamashina I (1978) Isolation and characterization of a mannan-binding protein from rabbit liver. Biochem Biophys Res Commun 81:1018
Kawasaki N, Kawasaki T, Yamashina I (1983) Isolation and characterization of a mannan-binding protein from human serum. J Biochem (Tokyo) 94:937
Sastry K, Herman GA, Day L et al (1989) The human mannose-binding protein gene. Exon structure reveals its evolutionary relationship to a human pulmonary surfactant gene and localization to chromosome 10. J Exp Med 170:1175
Guo N, Mogues T, Weremowicz S et al (1998) The human ortholog of rhesus mannose-binding protein-A gene is an expressed pseudogene that localizes to chromosome 10. Mamm Genome 9:246
Holmskov UL (2000) Collectins and collectin receptors in innate immunity. APMIS Suppl 100:1
Wallis R (2002) Structural and functional aspects of complement activation by mannose-binding protein. Immunobiology 205:433
Sheriff S, Chang CY, Ezekowitz RA (1994) Human mannose-binding protein carbohydrate recognition domain trimerizes through a triple alpha-helical coiled-coil. Nat Struct Biol 1:789
Jensen PH, Weilguny D, Matthiesen F et al (2005) Characterization of the oligomer structure of recombinant human mannan-binding lectin. J Biol Chem 280:11043
Teillet F, Dublet B, Andrieu JP et al (2005) The two major oligomeric forms of human mannan-binding lectin: chemical characterization, carbohydrate-binding properties, and interaction with MBL-associated serine proteases. J Immunol 174:2870
Lu JH, Thiel S, Wiedemann H et al (1990) Binding of the pentamer/hexamer forms of mannan-binding protein to zymosan activates the proenzyme C1r2C1s2 complex, of the classical pathway of complement, without involvement of C1q. J Immunol 144:2287
Jensenius JC (2002) Mannose-binding lectin. Lancet 359:82
Weis WI, Drickamer K, Hendrickson WA (1992) Structure of a C-type mannose-binding protein complexed with an oligosaccharide. Nature 360:127
Iobst ST, Wormald MR, Weis WI et al (1994) Binding of sugar ligands to Ca(2+)-dependent animal lectins. I. Analysis of mannose binding by site-directed mutagenesis and NMR. J Biol Chem 269:15505
Thiel S, Moller-Kristensen M, Jensen L et al (2002) Assays for the functional activity of the mannan-binding lectin pathway of complement activation. Immunobiology 205:446
Madsen HO, Garred P, Kurtzhals JA et al (1994) A new frequent allele is the missing link in the structural polymorphism of the human mannan-binding protein. Immunogenetics 40:37
Madsen HO, Garred P, Thiel S et al (1995) Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J Immunol 155:3013
Madsen HO, Satz ML, Hogh B et al (1998) Different molecular events result in low protein levels of mannan-binding lectin in populations from southeast Africa and South America. J Immunol 161:3169
Lipscombe RJ, Sumiya M, Hill AV et al (1992) High frequencies in African and non-African populations of independent mutations in the mannose binding protein gene. Hum Mol Genet 1:709
Sumiya M, Super M, Tabona P et al (1991) Molecular basis of opsonic defect in immunodeficient children. Lancet 337:1569
Steffensen R, Thiel S, Varming K et al (2000) Detection of structural gene mutations and promoter polymorphisms in the mannan-binding lectin (MBL) gene by polymerase chain reaction with sequence-specific primers. J Immunol Methods 241:33
Garred P, Larsen F, Madsen HO et al (2003) Mannose-binding lectin deficiency-revisited. Mol Immunol 40:73
Thiel S, Holmskov U, Hviid L et al (1992) The concentration of the C-type lectin, mannan-binding protein, in human plasma increases during an acute phase response. Clin Exp Immunol 90:31
Super M, Thiel S, Lu J et al (1989) Association of low levels of mannan-binding protein with a common defect of opsonisation. Lancet 2:1236
Thiel S, Frederiksen PD, Jensenius JC (2005) Clinical manifestations of mannan-binding lectin deficiency. Mol Immunol (in press)
Nuytinck L, Shapiro F (2004) Mannose-binding lectin: laying the stepping stones from clinical research to personalized medicine. Personalized Med 1:35
Boniotto M, Braida L, Baldas V et al (2005) Evidence of a correlation between mannose binding lectin and celiac disease: a model for other autoimmune diseases. J Mol Med 83:308
Ohlenschlaeger T, Garred P, Madsen HO et al (2004) Mannose-binding lectin variant alleles and the risk of arterial thrombosis in systemic lupus erythematosus. N Engl J Med 351:260
Peterslund NA, Koch C, Jensenius JC et al (2001) Association between deficiency of mannose-binding lectin and severe infections after chemotherapy. Lancet 358:637
Neth O, Hann I, Turner MW et al (2001) Deficiency of mannose-binding lectin and burden of infection in children with malignancy: a prospective study. Lancet 358:614
Koch A, Melbye M, Sorensen P et al (2001) Acute respiratory tract infections and mannose-binding lectin insufficiency during early childhood. JAMA 285:1316
Matsushita M, Fujita T (2002) The role of ficolins in innate immunity. Immunobiology 205:490
Le Y, Lee SH, Kon OL et al (1998) Human L-ficolin: plasma levels, sugar specificity, and assignment of its lectin activity to the fibrinogen-like (FBG) domain. FEBS Lett 425:367
Matsushita M, Endo Y, Taira S et al (1996) A novel human serum lectin with collagen- and fibrinogen-like domains that functions as an opsonin. J Biol Chem 271:2448
Akaiwa M, Yae Y, Sugimoto R (1999) Hakata antigen, a new member of the ficolin/opsonin p35 family, is a novel human lectin secreted into bronchus/alveolus and bile. J Histochem Cytochem 47:777
Yae Y, Inaba S, Sato H et al (1991) Isolation and characterization of a thermolabile beta-2 macroglycoprotein (‘thermolabile substance’ or ‘Hakata antigen’) detected by precipitating (auto) antibody in sera of patients with systemic lupus erythematosus. Biochim Biophys Acta 1078:369
The C, Le Y, Lee SH et al (2000) M-ficolin is expressed on monocytes and is a lectin binding to N-acetyl-d-glucosamine and mediates monocyte adhesion and phagocytosis of Escherichia coli. Immunology 101:225
Lu J, Teh C, Kishore U et al (2002) Collectins and ficolins: sugar pattern recognition molecules of the mammalian innate immune system. Biochim Biophys Acta 1572:387
Lynch NJ, Roscher S, Hartung T et al (2004) L-ficolin specifically binds to lipoteichoic acid, a cell wall constituent of Gram-positive bacteria, and activates the lectin pathway of complement. J Immunol 172:1198
Matsushita M, Kuraya M, Hamasaki N et al (2002) Activation of the lectin complement pathway by H-ficolin (Hakata antigen). J Immunol 168:3502
Matsushita M, Endo Y, Fujita T (2000) Cutting edge: complement-activating complex of ficolin and mannose-binding lectin-associated serine protease. J Immunol 164:2281
Krarup A, Thiel S, Hansen A et al (2004) L-ficolin is a pattern recognition molecule specific for acetyl groups. J Biol Chem 279:47513
Inaba S, Okochi K, Yae Y et al (1990) Serological studies of an SLE-associated antigen-antibody system discovered as a precipitation reaction in agarose gel: the HAKATA antigen-antibody system. Fukuoka Igaku Zasshi 81:284
Atkinson AP, Cedzynski M, Szemraj J (2004) L-ficolin in children with recurrent respiratory infections. Clin Exp Immunol 138:517
Arora M, Munoz E, Tenner AJ (2001) Identification of a site on mannan-binding lectin critical for enhancement of phagocytosis. J Biol Chem 276:43087
Neth O, Jack DL, Johnson M et al (2002) Enhancement of complement activation and opsonophagocytosis by complexes of mannose-binding lectin with mannose-binding lectin-associated serine protease after binding to Staphylococcus aureus. J Immunol 169:4430
Ikeda K, Sannoh T, Kawasaki N et al (1987) Serum lectin with known structure activates complement through the classical pathway. J Biol Chem 262:7451
Matsushita M, Fujita T (1992) Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J Exp Med 176:1497
Thiel S, Vorup-Jensen T, Stover CM et al (1997) A second serine protease associated with mannan-binding lectin that activates complement. Nature 386:506
Takahashi M, Endo Y, Fujita T et al (1999) A truncated form of mannose-binding lectin-associated serine protease (MASP)-2 expressed by alternative polyadenylation is a component of the lectin complement pathway. Int Immunol 11:859
Vorup-Jensen T, Petersen SV, Hansen AG et al (2000) Distinct pathways of mannan-binding lectin (MBL)- and C1-complex autoactivation revealed by reconstitution of MBL with recombinant MBL-associated serine protease-2. J Immunol 165:2093
Matsushita M, Endo Y, Hamasaki N et al (2001) Activation of the lectin complement pathway by ficolins. Int Immunopharmacol 1:359
Fujita T, Endo Y, Nonaka M (2004) Primitive complement system-recognition and activation. Mol Immunol 41:103
Fujita T, Matsushita M, Endo Y (2004) The lectin-complement pathway—its role in innate immunity and evolution. Immunol Rev 198:185
Lynch NJ, Khan SU, Stover CM et al (2005) Composition of the lectin pathway of complement in Gallus gallus: absence of mannan-binding lectin-associated serine protease-1 in birds. J Immunol 174:4998
Endo Y, Sato T, Matsushita M et al (1996) Exon structure of the gene encoding the human mannose-binding protein-associated serine protease light chain: comparison with complement C1r and C1s genes. Int Immunol 8:1355
Endo Y, Takahashi M, Nakao M et al (1998) Two lineages of mannose-binding lectin-associated serine protease (MASP) in vertebrates. J Immunol 161:4924
Schwaeble W, Dahl MR, Thiel S et al (2002) The mannan-binding lectin-associated serine proteases (MASPs) and MAp19: four components of the lectin pathway activation complex encoded by two genes. Immunobiology 205:455
Stover C, Endo Y, Takahashi M et al (2001) The human gene for mannan-binding lectin-associated serine protease-2 (MASP-2), the effector component of the lectin route of complement activation, is part of a tightly linked gene cluster on chromosome 1p36.2–3. Genes Immun 2:119
Stover CM, Lynch NJ, Hanson SJ et al (2004) Organization of the MASP2 locus and its expression profile in mouse and rat. Mamm Genome 15:887
Kuraya M, Matsushita M, Endo Y et al (2003) Expression of H-ficolin/Hakata antigen, mannose-binding lectin-associated serine protease (MASP)-1 and MASP-3 by human glioma cell line T98G. Int Immunol 15:109
Bork P, Beckmann G (1993) The CUB domain. A widespread module in developmentally regulated proteins. J Mol Biol 231:539
Romao MJ, Kolln I, Dias JM et al (1997) Crystal structure of acidic seminal fluid protein (aSFP) at 1.9 A resolution: a bovine polypeptide of the spermadhesin family. J Mol Biol 274:650
Gregory LA, Thielens NM, Matsushita M et al (2004) The X-ray structure of human MBL-associated protein 19 (MAp19) and its interaction site with mannan-binding lectin and L-ficolin. J Biol Chem 279:29391
Feinberg H, Uitdehaag JC, Davies JM et al (2003) Crystal structure of the CUB1-EGF-CUB2 region of mannose-binding protein associated serine protease-2. EMBO J 22:2348
Gregory LA, Thielens NM, Arlaud GJ et al (2003) X-ray structure of the Ca2+-binding interaction domain of C1s. Insights into the assembly of the C1 complex of complement. J Biol Chem 278:32157
Selander-Sunnerhagen M, Ullner M, Persson E et al (1992) How an epidermal growth factor (EGF)-like domain binds calcium. High resolution NMR structure of the calcium form of the NH2-terminal EGF-like domain in coagulation factor X. J Biol Chem 267:19642
Rao Z, Handford P, Mayhew M et al (1995) The structure of a Ca(2+)-binding epidermal growth factor-like domain: its role in protein–protein interactions. Cell 82:131
Handford PA, Mayhew M, Baron M et al (1991) Key residues involved in calcium-binding motifs in EGF-like domains. Nature 351:164
Kirkitadze MD, Barlow PN (2001) Structure and flexibility of the multiple domain proteins that regulate complement activation. Immunology 180:146
Barlow PN, Baron M, Norman DG et al (1991) Secondary structure of a complement control protein module by two-dimensional 1H NMR. Biochemistry 30:997
Harmat V, Gal P, Kardos J et al (2004) The structure of MBL-associated serine protease-2 reveals that identical substrate specificities of C1s and MASP-2 are realized through different sets of enzyme-substrate interactions. J Mol Biol 342:1533
Gaboriaud C, Rossi V, Bally I et al (2000) Crystal structure of the catalytic domain of human complement c1s: a serine protease with a handle. EMBO J 19:1755
Perona JJ, Craik CS (1997) Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J Biol Chem 272:29987
Krem MM, Di Cera E (2001) Molecular markers of serine protease evolution. EMBO J 20:3036
Krem MM, Prasad S, Di Cera E (2002) Ser(214) is crucial for substrate binding to serine proteases. J Biol Chem 277:40260
Budayova-Spano M, Lacroix M, Thielens NM et al (2002) The crystal structure of the zymogen catalytic domain of complement protease C1r reveals that a disruptive mechanical stress is required to trigger activation of the C1 complex. EMBO J 21:231
Ambrus G, Gal P, Kojima M et al (2003) Natural substrates and inhibitors of mannan-binding lectin-associated serine protease-1 and -2: a study on recombinant catalytic fragments. J Immunol 170:1374
Rossi V, Bally I, Thielens NM et al (1998) Baculovirus-mediated expression of truncated modular fragments from the catalytic region of human complement serine protease C1s. Evidence for the involvement of both complement control protein modules in the recognition of the C4 protein substrate. J Biol Chem 273:1232
Rossi V, Cseh S, Bally I et al (2001) Substrate specificities of recombinant mannan-binding lectin-associated serine proteases-1 and -2. J Biol Chem 276:40880
Freer ST, Kraut J, Robertus JD et al (1970) Chymotrypsinogen: 2.5-angstrom crystal structure, comparison with alpha-chymotrypsin, and implications for zymogen activation. Biochemistry 9:1997
Matsushita M, Matsushita A, Endo Y et al (2004) Origin of the classical complement pathway: Lamprey orthologue of mammalian C1q acts as a lectin. Proc Natl Acad Sci U S A 101:10127
Hernandez JF, Bersch B, Petillot Y et al (1997) Chemical synthesis and characterization of the epidermal growth factor-like module of human complement protease C1r. J Pept Res 49:221
Thielens NM, Enrie K, Lacroix M et al (1999) The N-terminal CUB-epidermal growth factor module pair of human complement protease C1r binds Ca2+ with high affinity and mediates Ca2+-dependent interaction with C1s. J Biol Chem 274:9149
Wallis R, Dodd RB (2000) Interaction of mannose-binding protein with associated serine proteases: effects of naturally occurring mutations. J Biol Chem 275:30962
Thielens NM, Cseh S, Thiel S et al (2001) Interaction properties of human mannan-binding lectin (MBL)-associated serine proteases-1 and -2, MBL-associated protein 19, and MBL. J Immunol 166:5068
Zundel S, Cseh S, Lacroix M et al (2004) Characterization of recombinant mannan-binding lectin-associated serine protease (MASP)-3 suggests an activation mechanism different from that of MASP-1 and MASP-2. J Immunol 172:4342
Arlaud GJ, Gaboriaud C, Thielens NM et al (2001) Structural biology of C1: dissection of a complex molecular machinery. Immunol Rev 180:136
Thiel S, Petersen SV, Vorup-Jensen T et al (2000) Interaction of C1q and mannan-binding lectin (MBL) with C1r, C1s, MBL-associated serine proteases 1 and 2, and the MBL-associated protein MAp19. J Immunol 165:878
Chen CB, Wallis R (2001) Stoichiometry of complexes between mannose-binding protein and its associated serine proteases. Defining functional units for complement activation. J Biol Chem 276:25894
Cseh S, Vera L, Matsushita M et al (2002) Characterization of the interaction between L-ficolin/p35 and mannan-binding lectin-associated serine proteases-1 and -2. J Immunol 169:5735
Wallis R, Shaw JM, Uitdehaag J et al (2004) Localization of the serine protease-binding sites in the collagen-like domain of mannose-binding protein: indirect effects of naturally occurring mutations on protease binding and activation. J Biol Chem 279:14065
Larsen F, Madsen HO, Sim RB et al (2004) Disease-associated mutations in human mannose-binding lectin compromise oligomerization and activity of the final protein. J Biol Chem 279:21302
Matsushita M, Ezekowitz RA, Fujita T (1995) The Gly-54–>Asp allelic form of human mannose-binding protein (MBP) fails to bind MBP-associated serine protease. Biochem J 311(Pt 3):1021
Chen CB, Wallis R (2004) Two mechanisms for mannose-binding protein modulation of the activity of its associated serine proteases. J Biol Chem 279:26058
Matsushita M, Thiel S, Jensenius JC et al (2000) Proteolytic activities of two types of mannose-binding lectin-associated serine protease. J Immunol 165:2637
Petersen SV, Thiel S, Jensenius JC (2001) The mannan-binding lectin pathway of complement activation: biology and disease association. Mol Immunol 38:133
Hajela K, Kojima M, Ambrus G et al (2002) The biological functions of MBL-associated serine proteases (MASPs). Immunobiology 205:467
Presanis JS, Hajela K, Ambrus G et al (2004) Differential substrate and inhibitor profiles for human MASP-1 and MASP-2. Mol Immunol 40:921
Petersen SV, Thiel S, Jensen L et al (2000) Control of the classical and the MBL pathway of complement activation. Mol Immunol 37:803
Gulati S, Sastry K, Jensenius JC et al (2002) Regulation of the mannan-binding lectin pathway of complement on Neisseria gonorrhoeae by C1-inhibitor and alpha 2-macroglobulin. J Immunol 168:4078
Moller-Kristensen M, Jensenius JC, Jensen L et al (2003) Levels of mannan-binding lectin-associated serine protease-2 in healthy individuals. J Immunol Methods 282:159
Pickering MC, Botto M, Taylor PR et al (2000) Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol 76:227
Carlsson M, Sjoholm AG, Eriksson L et al (2005) Deficiency of the mannan-binding lectin pathway of complement and poor outcome in cystic fibrosis: bacterial colonization may be decisive for a relationship. Clin Exp Immunol 139:306
Ytting H, Christensen IJ, Thiel S et al (2005) Serum mannan-binding lectin-associated serine protease 2 levels in colorectal cancer: relation to recurrence and mortality. Clin Cancer Res 11:1441
Ytting H, Christensen IJ, Jensenius JC et al (2005) Preoperative mannan-binding lectin pathway and prognosis in colorectal cancer. Cancer Immunol Immunother 54:265
Iwaki D, Fujita T (2005) Production and purification of recombinants of mouse MASP-2 and sMAP. J Endotoxin Res 11:47
Liu M, Endo Y, Iwaki D, Nakata M, Matsushita M, Wada I, Inoue K, Munakata M, Fujita T (2005) Human M-ficolin is a secretory protein that activates the lectin complement pathway. J Immunol 175:3150
Author information
Authors and Affiliations
Corresponding author
Additional information
Note added in proof
Recent results demonstrate M-ficolin to be present in secretory vesicles of myeloid cells including monocytes and polymorphnuclear and in type II lung epithelial cells [115]
Rights and permissions
About this article
Cite this article
Sørensen, R., Thiel, S. & Jensenius, J.C. Mannan-binding-lectin-associated serine proteases, characteristics and disease associations. Springer Semin Immun 27, 299–319 (2005). https://doi.org/10.1007/s00281-005-0006-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-005-0006-z