
Abstract
Purpose
Trifluridine/tipiracil (FTD/TPI) is approved for advanced colorectal and gastric/gastroesophageal cancer; however, data in patients with renal impairment (RI) are limited. This phase I study evaluated FTD/TPI in patients with advanced solid tumors and varying degrees of RI to develop dosing guidance.
Methods
Patients were enrolled into normal renal function (CrCl ≥ 90 mL/min), mild RI (CrCl 60–89 mL/min), or moderate RI (CrCl 30–59 mL/min) cohorts and administered the recommended FTD/TPI dose (35 mg/m2 twice daily, days 1–5 and 8–12; 28-day cycle). Based on interim pharmacokinetics/safety data, patients with severe RI (CrCl 15–29 mL/min) were enrolled and received FTD/TPI 20 mg/m2 twice daily.
Results
Forty-three patients (normal renal function [n = 12]; mild RI [n = 12]; moderate RI [n = 11]; severe RI [n = 8]) were enrolled and treated. At steady state, compared to values in patients with normal renal function, FTD area under the curve (AUC) was not significantly different in patients with RI, but TPI AUC was significantly higher and increased with RI severity. FTD/TPI safety profile was consistent with prior experience, but grade ≥ 3 adverse events (AEs) were more frequent in the RI cohorts (83.3% [mild], 90.9% [moderate], 75.0% [severe], and normal [50.0%]). Hematologic AEs (anemia and neutropenia) were more frequent with RI. Overall, seven patients discontinued because of unrelated, nonhematologic AEs.
Conclusion
FTD/TPI is safe and tolerable at the recommended 35 mg/m2 dose in patients with mild/moderate RI and at the reduced 20 mg/m2 dose in patients with severe RI.
Trial registration
NCT02301117, registration date: November 21, 2014.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Trifluridine and tipiracil (FTD/TPI) is an oral cytotoxic chemotherapy comprising trifluridine (FTD), a thymidine-based antineoplastic nucleoside analog, and tipiracil (TPI), a thymidine phosphorylase inhibitor, formulated at a molar ratio of 1.0:0.5 [1]. TPI significantly improves the oral bioavailability of FTD by inhibiting its degradation by thymidine phosphorylase, thereby increasing its systemic exposure [1, 2].
FTD/TPI demonstrated antitumor activity and tolerability in phase I–II clinical trials across a range of tumor types [3,4,5,6,7]. Pharmacokinetic analyses of FTD/TPI, including a mass balance study in patients with solid tumors, showed that following oral administration, both FTD and TPI are rapidly absorbed, with median times to maximum observed plasma concentration (Tmax) values ranging from 1 to 2 h, and are eliminated with a half-life of 1.5–4.0 h [4, 5, 8]. FTD is mainly eliminated via metabolism by thymidine phosphorylase to the inactive metabolite trifluoromethyluracil (FTY); < 3% of FTD was found unchanged in the urine/feces [8, 9]. This suggested that the main pathway of elimination of FTD is nonrenal. However, TPI is mostly eliminated via renal excretion; 27% of TPI was found unchanged in the urine [4, 5, 8, 9].
In the phase III RECOURSE and TAGS trials [10, 11], FTD/TPI demonstrated significantly improved survival versus placebo, and a tolerable safety profile in patients with treatment-refractory, metastatic colorectal, gastric, or gastroesophageal cancer. The most frequent adverse events (AEs) associated with FTD/TPI use were hematologic, including neutropenia and anemia. In both trials, FTD/TPI was dosed at 35 mg/m2 twice daily (BID) on days 1–5 and 8–12 of a 28-day cycle (administered orally within 1 h after a meal), with days 13–28 constituting the recovery period. Based on these results [10, 11], FTD/TPI was approved for both indications.
A significant proportion of patients with advanced solid tumors have renal impairment (RI) as defined by a creatinine clearance (CrCl) value < 90 mL/min [12,13,14]. RI alters drug pharmacokinetics not only by impairing urinary clearance but also by affecting drug absorption, metabolism, and distribution. Studies in cancer patients with RI have shown that the pharmacokinetics of many anticancer drugs are significantly altered by RI, and some patients experienced higher incidences of AEs associated with overexposure [12, 13]. These studies demonstrate the need for dosing adjustments of anticancer therapy in patients with RI. As TPI is primarily eliminated by urinary excretion, RI is expected to impact TPI excretion and increase its exposure, and consequently, increase FTD exposure. Patients with RI may require FTD/TPI dosing adjustments. However, data on FTD/TPI use in patients with RI are limited; the phase III clinical trials were not designed to assess the use of FTD/TPI in patients with RI, and patients with moderate to severe RI were underrepresented in these trials [10, 11]. Therefore, further research was needed to develop dosing recommendations for FTD/TPI in patients with RI.
This phase I study was conducted to evaluate the pharmacokinetics and safety of FTD/TPI in patients with advanced solid tumors and normal renal function or varying degrees of RI, and thereby develop guidance for making dosing adjustments in patients with RI.
Methods
Study design, treatment, and endpoints
This was a phase I open-label study (Clinicaltrials.gov registration: NCT02301117) in patients with advanced solid tumors and varying degrees of RI or normal renal function. This study consisted of 2 parts: a pharmacokinetic part (cycle 1) and an extension part (cycle 2 onwards; Supplementary Fig. S1). The estimated CrCl by the Cockroft–Gault equation was used to categorize patients into cohorts based on their degree of RI [15]. CrCl was tested within 7 days prior to starting the study medication, and patients on dialysis were excluded. This was not designed as a dose-finding study, and patients were enrolled based on baseline renal function. Patients were initially enrolled into one of three cohorts (cohorts 1–3) according to their baseline renal function at study entry: normal renal function (CrCl ≥ 90 mL/min), mild RI (CrCl 60–89 mL/min), or moderate RI (CrCl 30–59 mL/min). In the pharmacokinetic part, patients in cohorts 1–3 received the same FTD/TPI dose as patients in the phase III TAGS and RECOURSE trials (35 mg/m2 BID on days 1–5 and 8–12 of a 28-day cycle). Subgroup data from an earlier phase II study [16; data on file] and from the phase III RECOURSE trial [17] indicated tolerability of the 35 mg/m2 BID dose in patients with mild or moderate RI. Therefore, these patients received the recommended FTD/TPI dose. Study medication was administered orally, within 1 h after a meal.
Dose selection and enrollment in the severe RI cohort
An interim safety and pharmacokinetic assessment was conducted (cutoff date: December 21, 2015) after patients in cohorts 1–3 had completed cycle 1 (the pharmacokinetic part). Safety results obtained were consistent with previous observations [10, 11, 16,17,18; data on file]. Although treatment-related grade ≥ 3 AEs occurred more frequently in the moderate RI cohort (46%) than in the mild RI or the normal renal function cohort (17% each), the discontinuation rate after cycle 1 was ≈20% in each cohort and was not biased by RI. Pharmacokinetic data suggested an increase in FTD area under the curve from 0 to 12 h (AUC0–12) of approximately 15% and 50% in patients with mild and moderate impairment, respectively, compared with that in patients with normal renal function, indicating a correlation between FTD oral clearance (CL/F) and renal function. Regression analysis with the formula FTD CL/F = 0.3432 × CrCl0.4870 extrapolated the CL/F in patients with severe RI (CrCl 15–29 mL/min) to be approximately half that in patients with normal renal function. Based on pharmacokinetic analysis in cohorts 1–3, 20 mg/m2 BID was designated to have comparable exposure in patients with severe RI as the recommended 35 mg/m2 BID dose in patients with normal renal function and was considered to have an adequate safety margin. Therefore, the dose level for the fourth, severe RI cohort was chosen as 20 mg/m2 BID, on the same dosing schedule as the other cohorts. Initially, patients in the severe RI cohort were enrolled one at a time to ensure safety and tolerability. After the third patient had completed cycle 1, the remaining patients were enrolled in parallel. Patients in all cohorts who had completed cycle 1 (in the pharmacokinetic part of the study) were eligible to enter the extension part.
In the extension part, patients continued to receive FTD/TPI at the same dose and schedule as cycle 1 until disease progression, unacceptable toxicity (described later), or the patient’s request or physician’s decision to withdraw. The primary objectives of the study were: (1) to assess the safety and tolerability of FTD/TPI in patients with advanced solid tumors and varying degrees of RI and (2) to compare the plasma pharmacokinetic profile of FTD and TPI in patients with normal renal function to those with varying degrees of RI, to evaluate the impact of RI on the pharmacokinetic profile of FTD and TPI.
Patients
Adult patients with advanced solid tumors (except breast cancer), normal renal function or varying degrees of RI (as described above), and not on dialysis were eligible for enrollment. Other inclusion criteria included an Eastern Cooperative Oncology Group performance status (ECOG PS) of 2 or lower, ability to take oral medications, and adequate organ function (including a hemoglobin of ≥ 9.0 g/dL and absolute neutrophil count of ≥ 1.5 × 109/L). Key exclusion criteria were prior FTD/TPI treatment, serious illness (including brain metastases, active infection, and uncontrolled diabetes), major surgery, extended field radiation, or therapy with an investigational agent ≤ 4 weeks of the treatment start date.
This study was conducted in accordance with the Declaration of Helsinki and good clinical practice guidelines, and approval was obtained from each institution’s review board or ethics committee. All patients provided written informed consent prior to participation in the trial.
Safety assessments and criteria for dosing modifications or treatment discontinuation
AEs were recorded from the time of enrollment into the study until 30 days after the last study drug dose and were graded according to National Cancer Institute Common Terminology Criteria version 4.03.
In case of toxicity, a maximum of 3 dose reductions (by 5 mg/m2 per dose) were permitted to a minimum of 20 mg/m2 BID for patients in the normal renal function and mild and moderate RI cohorts and a maximum of 1 dose reduction (by 5 mg/m2 per dose) down to 15 mg/m2 was permitted for patients in the severe RI cohort.
Patients with uncomplicated grade ≤ 3 hematologic toxicities (neutropenia or thrombocytopenia) did not require dose reductions. The FTD/TPI dose was held if neutrophil count was < 500/mm3 or platelet count was < 50,000/mm3. For all patients with decreases in neutrophil and/or platelet counts, the next cycle of study treatment was not started until the resumption criteria (a neutrophil count of ≥ 1500/mm3 or a platelet count of ≥ 75,000/mm3) were met. Those patients with complicated grade ≥ 3 neutropenia or thrombocytopenia were administered supportive medications (hematopoietic growth factors) or were considered for a dose reduction in the next cycle, or both, depending on the severity of the event. Patients who experienced grade 4 neutropenia or thrombocytopenia that resulted in a > 1-week delay of the start of the next cycle were started at a reduced dose in the next cycle.
In case of nonhematologic toxicities, dosing was interrupted for the first three occurrences of grade ≥ 3 AEs and was resumed (once the AE resolved to baseline grade) at the next reduced dose level in the next dosing cycle. At the fourth occurrence of the grade ≥ 3 event, treatment was discontinued.
FTD/TPI treatment was permanently discontinued for one or more of the following reasons: an irreversible, treatment-related, grade 4 clinically relevant event; the fourth occurrence of a treatment-related, grade 3, nonhematologic AE; or an AE that required more than 3 dose reductions of FTD/TPI, a dose reduction below 15 mg/m2 BID for patients with severe RI, or a maximum dose delay of > 28 days from the scheduled start date of the next cycle.
Pharmacokinetic assessments
Blood samples were collected on days 1 and 12 of cycle 1 for measurement of plasma concentrations of FTD/TPI within 30 min prior to the dose (0 h), and 0.5, 1, 2, 4, 6, 8, 10, and 12 h after dose. Urine samples were collected prior to administration of the morning dose, and from 0 through 12 h after dose. The concentrations of FTD, FTY, and TPI in plasma and urine were measured using validated liquid chromatography-tandem mass spectrometry methods, as previously described [19]. The range of quantification for FTD was 5.00–5000 ng/mL, and twofold and fivefold dilutions were accurately quantitated. The range of quantification for TPI was 0.200–200 ng/mL. The accuracy values (relative error) for within-day and between-day reproducibility were − 4.0% to 3.0% and − 4.0% to 2.4%, respectively, for FTD, and − 3.0% to 3.8% and 1.0 to 4.0%, respectively, for TPI.
Pharmacokinetic endpoints and analyses
All pharmacokinetic data were analyzed using the Phoenix WinNonlin (Version 7; Certara LP, Princeton, NJ, USA) software. Noncompartmental methods using actual sampling times were applied for calculating the following plasma pharmacokinetic parameters for FTD and TPI: maximum observed plasma concentration (Cmax); area under the plasma concentration–time curve from time 0 to the last measurable plasma concentration estimated by the linear trapezoidal rule (AUC0–last), to infinity calculated for day 1 (AUC0–inf), and to end of dosing interval for day 12 only (AUCtau); apparent terminal phase elimination half-life (t1/2); terminal elimination oral clearance for day 1 (CL/F); steady-state oral clearance for day 12 (CLss/F); and apparent volume of distribution (Vd/F). The accumulation ratio of Cmax (RCmax) was calculated as the ratio of Cmax (day 12)/Cmax (day 1) and the accumulation ratio of AUC0–last (RAUC0–last) was calculated as the ratio of AUC0–last (day 12)/AUC0–last (day 1).
To determine an appropriate dose for patients with severe RI, a regression analysis between actual CrCl and CL/F values on Day 12 collected from patients with normal renal function, and mild and moderate RI was performed using the power model. According to the following power formula, CL/F at each RI level was calculated, and then the dose for patients with severe RI to provide the similar AUC with that for patients with normal renal function was estimated. The regression analysis was performed by the Taiho pharmacokinetic team.
Urine pharmacokinetic endpoints and parameters are described in Supplementary Methods.
Statistical analysis
Sample size considerations were based on the typical sample size (6–8 patients per cohort) recommended for RI studies [20]. Accounting for the individual variability in pharmacokinetic parameters (with a maximum covariance of 50%) and assuming a 25% dropout rate, it was estimated that for each cohort, 12 patients would need to be enrolled to obtain 9 evaluable patients. In the severe RI cohort, data from six evaluable patients were considered adequate for pharmacokinetic analysis.
The safety population comprised all patients who received at least 1 dose of FTD/TPI, and the pharmacokinetic analysis population comprised patients in the safety population with at least one concentration measurement on day 1, day 12, or both.
Plasma concentrations of FTD or TPI below the lower limit of quantification (LLOQ) in the predose samples or in early time points were set at 0. The collection time of the predose sample was set to 0, and actual postdose sampling times were expressed relative to the time of dosing. Plasma concentrations below the LLOQ in terminal samples or in the middle of the curve flanked by measurable concentrations were treated as missing values.
Effect of RI on FTD/TPI pharmacokinetics
Plasma pharmacokinetic parameters (Cmax, AUC0-inf, AUC0-last, and AUCtau) were analyzed by one-way analysis of variance (one-way ANOVA) using the categorical RI groups as class variables after the parameters were log-transformed, and comparisons were made between the normal renal function group and each of the RI groups. Point estimates of geometric mean ratios (GMRs) with their corresponding 90% confidence intervals (CIs) were constructed and back-transformed from the log-scale to express the estimates as ratios (each RI group to the control group). All analyses were carried out with Phoenix WinNonlin® software (Version 7.0, Certara LP; Princeton, New Jersey, USA) or SAS® statistical software, version 9.3 or a later version.
Results
Patient population
A total of 43 patients (12 patients with normal renal function, and 12, 11, and 8 patients with mild, moderate, and severe RI, respectively) were enrolled across 15 sites in the United States, Serbia, and the Czech Republic from March 2015 to November 2018. All patients received at least 1 dose of study drug. At data cutoff (January 31, 2019), 42 patients (98%) had discontinued (1 during the pharmacokinetic part, 7 prior to entering the extension part, and 34 during the extension part). Most discontinuations were due to disease progression (31 patients; 72%) or withdrawal of consent (6 patients; 14%).
Most patients were white (33/43; 76.7%); the median age was 64.0 years, and most patients (38/43; 88.4%) had gastrointestinal cancers (colorectal [n = 22], liver [n = 6], pancreas [n = 5], gastric [n = 3], and appendix [n = 2]). Median CrCl was 105.7 mL/min in the normal renal function cohort, and 70.1, 43.1, and 24.4 mL/min in the mild, moderate, and severe RI cohorts, respectively (Table 1). Mean baseline albumin was comparable across cohorts, ranging from 35.4 to 38.3 g/L. Nearly all patients (41/43; 95.3%) had received prior anticancer therapy, with 58.1% having received ≥ 3 prior regimens.
Pharmacokinetics
Among 41 patients who were included in the pharmacokinetic analysis, pharmacokinetic concentration data were available for 40 patients on day 1 of cycle 1, 38 patients on day 12 of cycle 1, and 37 patients at both time points. The reasons for the exclusion of patients from the pharmacokinetic analysis are listed in Supplementary Table S1.
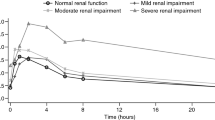
Mean plasma concentration–time profiles of FTD and TPI are shown in Figs. 1, 2, respectively, after the administration of the first dose (day 1, cycle 1) or multiple doses (day 12, cycle 1). On day 1, cycle 1, the mean plasma concentrations of FTD were comparable in the normal renal function, mild RI, and moderate RI cohorts, but were lower in the severe RI cohort. On day 12, cycle 1, the mean FTD concentration was lower in the normal renal function cohort than in the RI cohorts. The mean TPI plasma concentration was lowest in the normal renal function cohort and increased with the severity of RI on both days 1 and 12 of cycle 1.

Mean plasma concentration–time profile of FTD (pharmacokinetic population) on A day 1 and B day 12 of cycle 1. FTD trifluridine, SE standard error. Note: the lower limit for concentration in the log-linear scale is 0.01 ng/mL. Standard error bar values lower than 0.01 ng/mL are shown at 0.01 ng/mL

Mean plasma concentration–time profile of TPI (pharmacokinetic population) on A day 1 and B day 12 of cycle 1. SE standard error, TPI tipiracil. Note: the lower limit for concentration in the log-linear scale is 0.01 ng/mL. Standard error bar values lower than 0.01 ng/mL are shown at 0.01 ng/mL
Following the first dose of FTD/TPI (day 1, cycle 1), mean AUC values of FTD were similar between the normal renal function and the mild/moderate RI cohorts but was significantly lower in the severe RI cohort than the normal renal function cohort, which was consistent with the lower dose administered (− 51% for mean AUC0–last [GMR, 0.49; 90% CI, 0.33–0.73]; − 50% for mean AUC0–inf [GMR, 0.50; 90% CI, 0.33–0.76]; all P values < 0.05; Tables 2, 3). After multiple doses of FTD/TPI (day 12, cycle 1), compared with the normal renal function cohort, FTD AUC values were nearly unchanged in the mild cohort (by − 1% for both AUC0–last and AUCtau), and somewhat increased in the moderate and severe RI cohorts (by + 56% for AUC0–last and AUCtau for the moderate RI cohort; + 37% [AUC0–last] and + 45% [AUCtau] for the severe RI cohort), but these differences were not statistically significant (all P values > 0.05; Table 3). Mean accumulation ratios of AUC0–last increased with increasing RI (ranging from 2.80 to 6.09) on day 12 (Table 2).
FTD Cmax values followed a similar trend. After the first dose, mean FTD Cmax values were similar in the normal renal function, mild RI, and moderate RI cohorts but were somewhat decreased in the severe RI cohort (− 38%; GMR, 0.62), consistent with the lower dosing in this cohort. Following multiple doses, the differences in Cmax values between the normal renal function and RI cohorts were not significant; GMRs ranged from 0.84 to 1.19 (all P values > 0.05) on day 12 (Table 3).
Mean CL/F and CLss/F values for FTD showed the opposite trend to AUC values. Whereas mean CL/F values were similar across cohorts on day 1, a decreasing trend in mean CLss/F was noted with increasing RI on day 12 (Table 2). On both days, mean clearance values were not significantly different between the normal renal function and RI cohorts (Table 3).
On the other hand, TPI plasma exposures significantly increased with the severity of RI on both days 1 and 12 of cycle 1 and were lowest in the normal renal function cohort. On day 1, mean AUC0–last values were increased by + 65% (GMR, 1.65; 90% CI, 1.16–2.36; P < 0.05), + 121% (GMR, 2.21; 90% CI, 1.54–3.18; P < 0.05), and + 224% (GMR, 3.24; 90% CI, 2.15–4.89; P < 0.05) in the mild RI, moderate RI, and severe RI cohorts, respectively, compared with the normal renal function cohort. After multiple doses on day 12, mean AUC0–last values were increased by + 42% (GMR, 1.42; 90% CI, 1.00–2.03), + 139% (GMR, 2.39; 90% CI, 1.65–3.47; P < 0.05), and + 308% (GMR, 4.08; 90% CI, 2.66–6.26) in the mild RI, moderate RI, and severe RI cohorts, respectively, compared with the normal renal function cohort (Table 3). Trends in AUCinf and AUCtau were consistent with AUC0–last, although these values were not calculable in almost all patients in the severe RI cohort (Tables 2, 3). No marked accumulation in AUC0–last was observed in TPI on day 12 (Table 2). Similar to AUC values, mean Cmax values for TPI were significantly higher in the RI cohorts than in the normal renal function cohort on both day 1 (ranging from + 91% to + 113%) and day 12 (ranging from + 51% to + 158%), and these values increased with the severity of RI (Tables 2, 3).
Mean CL/F (day 1, cycle 1) and CLss/F values (day 12, cycle 1) for TPI were significantly lower in the mild and moderate RI cohorts than in the normal renal function cohort; GMRs were 0.66 (P = 0.0748) and 0.48 for CL/F and 0.65 and 0.48 for CLss/F (all the other P values < 0.05; Table 3). Mean clearance was not calculable for most patients in the severe RI cohort (Table 2).
Urine pharmacokinetic findings for FTD and TPI are shown in Supplementary Table S2 and are described in the Supplement.
Safety
All enrolled patients received at least one dose of FTD/TPI and were evaluable for safety (N = 43). The median treatment duration was 60 days (range, 1–306) for the overall population (42.0, 78.0, 71.0, and 52.5 days for the normal renal function and mild, moderate, and severe RI cohorts, respectively). Median relative dose intensity (ratio of delivered to planned) was 86.0% for the overall population (89.8%, 85.2%, 83.2%, and 91.0% for the various cohorts), and the median number of cycles completed were similar (2.0; range, 2.0–3.0) across cohorts (Supplementary Table S3).
Overall, 37 patients (86.0%) experienced a treatment-related AE, and 32 patients (74.4%) had a grade ≥ 3 AE of any causality. The overall incidence of any-cause grade ≥ 3 AEs was higher in the RI cohorts (83.3%, 90.9%, and 75.0% in the mild, moderate, and severe RI cohorts, respectively) than in the normal renal function cohort (50.0%); incidences of most grade ≥ 3 AEs were comparable in the severe and mild RI cohorts (Table 4 and Supplementary Table S4). The incidence of serious AEs was higher in the mild (58.3%) and moderate RI cohorts (45.5%) than in the normal renal function cohort (33.3%). Overall, the most common serious AEs were sepsis (n = 4), acute renal failure (n = 3), and small-intestinal obstruction and pneumonia (n = 2, each).
Overall, 5 patients (1 with mild RI, 2 with moderate RI, and 2 with severe RI) had one dose reduction each. In 4 patients, the dose reductions were reported during cycle 2, and one patient in the severe RI cohort had a dose reduction during cycle 9. Seven patients (16.3%) discontinued because of AEs, none of which were related to treatment. These discontinuations included 1 patient in the normal renal function cohort (due to decreased appetite), 3 patients in the mild RI cohort (due to abdominal pain, insomnia, and acute renal failure; n = 1 each), 2 patients in the moderate RI cohort (both due to small-intestinal obstruction), and 1 patient in the severe RI cohort (dehydration, fatigue, muscular weakness, and dyspnea). There were no deaths during the study.
In all patients, the most common AEs of any cause and grade were fatigue (55.8%), decreased appetite (48.8%), nausea (48.8%), anemia (39.5%), vomiting (34.9%), dehydration (27.9%), and diarrhea and dyspnea (25.6% each). Grade ≥ 3 AEs occurring in ≥ 10% of all patients were anemia and decreased neutrophil count (16.3% each) and neutropenia (11.6%). The only notable difference in AE incidences between the RI cohorts and the normal cohort pertained to anemia and neutropenia, which trended higher in the mild RI (any-grade anemia, 25.0%; any-grade neutropenia, 16.7%), moderate RI (72.7% and 27.3%, respectively), and severe RI cohorts (50.0% and 25.0%, respectively) than in the normal renal function cohort (16.7% and 8.3%, respectively). Grade ≥ 3 hematologic AEs led to dose interruptions in 1, 5, and 2 patients in the normal renal function, mild RI, and moderate RI cohorts, respectively; no dose interruptions due to hematologic toxicities were reported in the severe RI cohort. No patients had dose reductions due to grade ≥ 3 hematologic toxicities. Overall, no treatment discontinuations due to hematologic toxicities were reported.
Discussion
In view of the limited data available for FTD/TPI activity/safety in patients with RI, this phase I study was designed to provide guidance for FTD/TPI dosing in patients with advanced tumors and varying degrees of RI. In this study, patients with mild and moderate RI received 35 mg/m2 BID (on days 1–5 and 8–12 of a 28-day cycle), the recommended dose for patients with normal renal function, based on safety data in earlier phase II and III trials showing tolerability in patients with mild/moderate RI [16,17,18; data on file]. Based on the interim pharmacokinetic and safety data obtained from cohorts 1–3 in cycle 1, the dose for the severe RI cohort was designated as 20 mg/m2 BID (on days 1–5 and 8–12 of a 28-day cycle).
Given that absorbed TPI is mainly eliminated by urinary excretion [4, 5, 8], RI was expected to increase TPI exposure and thereby, enhance FTD exposure. In this study, the evaluation of FTD’s and TPI’s pharmacokinetic parameters in patients with RI or normal renal function indicated a significant effect of RI on TPI exposures. However, the effect of mild or moderate RI on FTD exposures was relatively marginal. Following administration of the first dose, FTD plasma concentrations and exposures were lower in the severe RI cohort than in the normal cohort, which reflected the lower dose that was administered. At steady state, no significant differences were observed in FTD plasma exposures between patients with RI and those in the normal cohort, although FTD plasma exposures showed a trend of increase with increased RI (mean accumulation ratio of AUC0–last in the severe RI cohort was nearly twofold that in the normal renal function cohort). It is to be noted that the steady-state plasma exposures of FTD in patients in the severe RI cohort who received FTD/TPI at a lower starting dose (20 mg/m2 BID) were similar to those observed in patients in the mild and moderate RI cohorts who received FTD/TPI at 35 mg/m2 BID. In contrast to FTD, significant increases in TPI plasma exposures were observed in all RI cohorts compared with the normal renal function cohort, both after administration of the first dose and at steady state. Plasma exposures of TPI (Cmax and AUCs) increased with the severity of RI, and the mean clearance decreased with severity. However, TPI accumulation ratios (RAUC0–last and RCmax) did not increase appreciably with RI.
RI may also affect the pharmacokinetics of chemotherapeutic agents through other indirect effects on nonrenal clearance, including absorption or hepatic metabolism [12, 13]. However, an earlier study evaluating FTD/TPI in patients with varying degrees of hepatic impairment showed that FTD and TPI exposures were not increased by hepatic impairment [18], which suggested that changes in nonrenal clearance may have a limited impact on exposure of FTD or TPI when administered together as FTD/TPI.
Study treatment exposure (median number of initiated/completed cycles; relative dose intensity) was comparable across cohorts, indicating that renal function did not have an impact on treatment compliance. The overall safety profile of FTD/TPI observed in this study was consistent with previous observations, and no new safety concerns were observed, which suggested that the increased TPI exposures did not impact the safety profile of FTD/TPI. Study discontinuation was overwhelmingly related to disease progression, not toxicity, and no AE-related deaths occurred. In patients with mild or moderate RI, FTD/TPI at 35 mg/m2 BID resulted in a higher frequency of grade ≥ 3 toxicities and serious AEs than in those with normal renal function, although incidences of any-grade treatment-related AEs were not increased in these cohorts. There were no clinically meaningful differences in the FTD/TPI safety profile between patients with severe RI who received an adjusted 20 mg/m2 BID dose and those with mild or moderate RI who received the regular dose, indicating that this reduced dose was tolerable in patients with severe RI. The frequency of grade ≥ 3 hematologic AEs, including anemia and neutropenia, trended higher in patients with RI than in those with normal renal function, although the small numbers of patients precluded any real comparison of AE rates between the cohorts. The increased incidence of grade ≥ 3 anemia in patients with severe RI could be partly explained by the decreased erythropoietin production and impaired iron absorption in these patients [21], which predispose them to anemia. Heme-related AEs were managed with dose interruptions (which were more frequent in the mild RI cohort), and no permanent discontinuations of treatment due to heme-related AEs were reported across cohorts.
These observations are consistent with results from post hoc subgroup analyses of FTD/TPI safety in patients with mild or moderate RI in the phase III RECOURSE and TAGS trials [17, 18]. In both trials, the rates of hematologic toxicities (in particular, neutropenia and anemia) were higher in patients with mild or moderate RI. However, these AEs were managed with dosing modifications and supportive medications. Nearly all cases of grade ≥ 3 neutropenia resolved; median times to resolution were similar in patients with normal renal function and in those with RI [18]. Overall, these data indicate that heme-related AEs can be monitored and managed in patients with RI without the need for treatment discontinuation.
One important limitation in this study was the small sample sizes of the cohorts. Although this sample size was sufficient to study the effects of RI on FTD/TPI pharmacokinetics, robust comparisons of safety could not be performed. In addition, plasma protein unbound fractions of FTD and TPI were not quantitated. Given that FTD mainly binds to albumin [9] and albumin is a significant covariate of FTD clearance [22], the effect of hypoalbuminemia, including that of ongoing renal dysfunction on the evaluation of FTD pharmacokinetics, could not be ruled out. However, in this study, baseline albumin levels were comparable between the normal renal function and the RI cohorts. In addition, there were no clinically meaningful changes in albumin or other lab parameters from baseline to the last study visit in any cohort, and there were no renal function-dependent changes in these parameters, which indicated the absence of significant worsening of renal function during the study. Therefore, it is unlikely that the evaluation of FTD/TPI pharmacokinetics was affected by changes in albumin levels or worsening of renal function.
In conclusion, the results of this study indicate that FTD/TPI at the recommended dose of 35 mg/m2 BID was tolerable in patients with advanced solid tumors and mild or moderate RI, with no unexpected safety concerns noted. Even with increased TPI exposures, FTD steady-state plasma exposures in patients with RI were comparable to those in patients with normal renal function. Although these patients may receive the recommended dose without the need for dosing adjustments, they should be closely monitored for toxicity, particularly hematologic toxicity. The reduced FTD/TPI starting dose of 20 mg/m2 BID was found to be tolerable and appropriate for patients with advanced solid tumors and severe RI, and no meaningful changes in safety were noted between the severe RI cohort at the adjusted dose and the mild or moderate RI cohorts at the recommended dose. On the basis of these results, the FTD/TPI dosage for patients with severe RI has been recommended as 20 mg/m2 BID on days 1–5 and days 8–12 of each 28-day cycle, allowing for a dose reduction to 15 mg/m2 in case of intolerability [9].
Availability of data and material
Data generated or analyzed during this study are on file with Taiho Oncology, Inc., and Taiho Pharmaceuticals Co., Ltd., and are not publicly available. Inquiries about data access should be sent to th-datasharing@taiho.co.jp.
Code availability
Phoenix WinNonlin® software (Version 7.0, Certara LP; Princeton, New Jersey, USA) or SAS® statistical software, version 9.3 or a later version.
References
Emura T, Murakami Y, Nakagawa F, Fukushima M, Kitazato K (2004) A novel antimetabolite, TAS-102 retains its effect on FU-related resistant cancer cells. Int J Mol Med 13(4):545–549
Cleary JM, Rosen LS, Yoshida K, Rasco D, Shapiro GI, Sun W (2017) A phase 1 study of the pharmacokinetics of nucleoside analog trifluridine and thymidine phosphorylase inhibitor tipiracil (components of TAS-102) vs trifluridine alone. Invest New Drugs 35(2):189–197
Bendell JC, Rosen LS, Mayer RJ et al (2015) Phase 1 study of oral TAS-102 in patients with refractory metastatic colorectal cancer. Cancer Chemother Pharmacol 76(5):925–932
Doi T, Ohtsu A, Yoshino T et al (2012) Phase I study of TAS-102 treatment in Japanese patients with advanced solid tumours. Br J Cancer 107(3):429–434
Hong DS, Abbruzzese JL, Bogaard K et al (2006) Phase I study to determine the safety and pharmacokinetics of oral administration of TAS-102 in patients with solid tumors. Cancer 107(6):1383–1390
Overman MJ, Varadhachary G, Kopetz S et al (2008) Phase 1 study of TAS-102 administered once daily on a 5-day-per-week schedule in patients with solid tumors. Invest New Drugs 26(5):445–454
Scagliotti G, Nishio M, Satouchi M et al (2016) A phase 2 randomized study of TAS-102 versus topotecan or amrubicin in patients requiring second-line chemotherapy for small cell lung cancer refractory or sensitive to frontline platinum-based chemotherapy. Lung Cancer 100:20–23
Lee JJ, Seraj J, Yoshida K et al (2016) Human mass balance study of TAS-102 using (14)C analyzed by accelerator mass spectrometry. Cancer Chemother Pharmacol 77(3):515–526
Taiho Oncology (2019) LONSURF (trifluridine and tipiracil) tablets, for oral use [prescribing information]. Taiho Oncology Inc, Princeton, NJ
Mayer RJ, Van Cutsem E, Falcone A et al (2015) Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med 372(20):1909–1919
Shitara K, Doi T, Dvorkin M et al (2018) Trifluridine/tipiracil versus placebo in patients with heavily pretreated metastatic gastric cancer (TAGS): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 19(11):1437–1448
Launay-Vacher V, Janus N, Deray G (2016) Renal insufficiency and cancer treatments. ESMO Open 1(4):e000091
Launay-Vacher V, Oudard S, Janus N et al (2007) Prevalence of renal insufficiency in cancer patients and implications for anticancer drug management: the renal insufficiency and anticancer medications (IRMA) study. Cancer 110(6):1376–1384
National Kidney Foundation (2002) K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 39(2 Suppl 1):S1-266
US FDA Guidance Document (2010) Pharmacokinetics in patients with impaired renal function: study design, data analysis and impact on dosing and labeling. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pharmacokinetics-patients-impaired-renal-function-study-design-data-analysis-and-impact-dosing-and. Accessed 27 April 2021
Yoshino T, Mizunuma N, Yamazaki K et al (2012) TAS-102 monotherapy for pretreated metastatic colorectal cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol 13(10):993–1001
Mayer RJ, Ohtsu A, Yoshino T et al (2016) Efficacy and safety results in patients with impaired renal and hepatic function in the RECOURSE trial. J Clin Oncol 34(15_suppl):3547
George B, Cutsem EV, Hochster HS et al (2020) Analysis of hematologic adverse events (HeAEs) in trifluridine/tipiracil (FTD/TPI)-treated patients (pts) with or without renal/hepatic impairment (RI/HI): Pooled safety analyses from TAGS and RECOURSE. J Clin Oncol 38(4_suppl):145
Saif MW, Rosen L, Rudek MA et al (2019) Open-label study to evaluate trifluridine/tipiracil safety, tolerability and pharmacokinetics in patients with advanced solid tumours and hepatic impairment. Br J Clin Pharmacol 85(6):1239–1246
Ibrahim S, Honig P, Huang SM, Gillespie W, Lesko LJ, Williams RL (2000) Clinical pharmacology studies in patients with renal impairment: past experience and regulatory perspectives. J Clin Pharmacol 40(1):31–38
Honda H, Hosaka N, Ganz T, Shibata T (2019) Iron metabolism in chronic kidney disease patients. Contrib Nephrol 198:103–111
Cleary JM, Mayer RJ, Van Cutsem E et al (2015) Population pharmacokinetic (PK) analysis of TAS-102 in patients (pts) with metastatic colorectal cancer (mCRC): results from 3 phase 1 trials and the phase 3 RECOURSE trial. J Clin Oncol 33(15_suppl):2579
Acknowledgements
This study was sponsored by Taiho Oncology, Inc., and Taiho Pharmaceutical Co., Ltd. The authors acknowledge Dr. Kenichiro Yoshida for the study design and regression model analysis. Medical writing and editorial assistance were provided by Vasupradha Vethantham, PhD, and Jennifer Robertson, PhD, of Ashfield Healthcare Communications (Lyndhurst, NJ, USA) and funded by Taiho Oncology, Inc.
Funding
This study was funded by Taiho Oncology, Inc., and Taiho Pharmaceutical Co. Ltd.
Author information
Authors and Affiliations
Contributions
M.W.S., L.R., I.Y., K.H., and Y.H. were involved in the study design. M.W.S., C.R.B., M.G.F., W.S., L.P., S.K., T.J.G., M.A.R., D.R.S., J.S., V.S., B.Z., and L.R. contributed to data collection. M.W.S., L.R., I.Y., K.A.B., K.H., and Y.H were involved in data analysis and/or interpretation. All authors meet the criteria for authorship as recommended by the International Committee of Medical Journal Editors. All authors take full responsibility for the scope, direction, content of, and editorial decisions relating to the manuscript, were involved at all stages of development, and have approved the submitted manuscript.
Corresponding author
Ethics declarations
Conflict of interest
M.W.S. reports research funding from Incyte, Lexicon, NGM Bio, Nouscom, Tesaro, and Yivia. C.R.B. is part of the speaker bureau for Taiho Oncology Inc. M.G.F. received research grant funding (to institution) from Amgen, AstraZeneca, and Novartis; has served on the advisory board of Amgen, Array Biopharma, Bayer, and Pfizer; is part of the speakers bureau for Amgen and Guardant360, and reports honoraria from Amgen. M.A.R. received research funding from Taiho Oncology, Inc, during the conduct of the study and from Celgene Corporation and the National Cancer Institute, outside the submitted work. K.H., I.Y., K.A.B., and Y.H are employees of Taiho Oncology, Inc. K.A.B. is a former employee and a stockholder of Eli Lilly. L.R. received research funding from Taiho Oncology, Inc. W.S., L.P., S.K., T.J.G., D.R.S., J.S., V.S., and B.Z have no financial disclosures to report.
Ethics approval
This study was performed in line with the Declaration of Helsinki. Approval was granted from the review board or ethics committee of each institution.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Saif, M.W., Becerra, C.R., Fakih, M.G. et al. A phase I, open-label study evaluating the safety and pharmacokinetics of trifluridine/tipiracil in patients with advanced solid tumors and varying degrees of renal impairment. Cancer Chemother Pharmacol 88, 485–497 (2021). https://doi.org/10.1007/s00280-021-04308-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-021-04308-z