
Abstract
To resolve the topological problems that threaten the function and structural integrity of nuclear and mitochondrial genomes and RNA molecules, human cells encode six different DNA topoisomerases including type IB enzymes (TOP1 and TOP1mt), type IIA enzymes (TOP2α and TOP2β) and type IA enzymes (TOP3α and TOP3β). DNA entanglements and the supercoiling of DNA molecules are regulated by topoisomerases through the introduction of transient enzyme-linked DNA breaks. The covalent topoisomerase–DNA complexes are the cellular targets of a diverse group of cancer chemotherapeutics, which reversibly stabilize these reaction intermediates. Here we review the structure–function and catalytic mechanisms of each family of eukaryotic DNA topoisomerases and the topoisomerase-targeting agents currently approved for patient therapy or in clinical trials, and highlight novel developments and challenges in the clinical development of these agents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
History
In 1971, James C. Wang discovered a new class of enzyme from Escherichia coli that catalyzes alterations in the topology of duplex DNA molecules, which are now referred to as DNA topoisomerases. The enzyme he described (protein ϖ or prokaryotic DNA topoisomerase I) had the ability to resolve negative superhelical turns (or underwinding of DNA strands) within closed circular, duplex DNA without the necessity of an energy cofactor [1]. He hypothesized a reaction mechanism of concerted DNA strand nicking and religation mediated by the formation of an enzyme–DNA covalent intermediate. In 1972, Champoux and Dulbecco reported a similar enzymatic activity from mouse embryo cells that was also able to relax DNA superhelical turns and later demonstrated the covalent linkage of the enzyme to a single nicked DNA strand by a phosphodiester bond to tyrosine [2, 3]. As with prokaryotic DNA topoisomerase I, the formation of a covalent enzyme–DNA intermediate obviated the need for an energy cofactor in the DNA nicking/closing reaction cycle of the DNA. It took almost 30 years before Pommier and co-workers identified a mitochondrial-targeted isoform of Champoux and Dulbecco’s eukaryotic TOP1, TOP1mt, which is only found in vertebrates [4].
In 1976, Gellert and colleagues isolated DNA gyrase, a distinct class of DNA topoisomerase from E. coli. This enzyme was able to introduce negative supercoils into DNA; however, the reaction required the cofactors ATP and Mg2+ [5]. Subsequent studies defined the formation of transient, protein-linked double-strand DNA breaks [6]. In 1980, Liu and Alberts [7] coined the term type II DNA topoisomerase to describe enzymes such as DNA gyrase or bacteriophage T4 topoisomerase that alter DNA topology by reversibly breaking both strands of duplex DNA. A eukaryotic type II DNA topoisomerase (TOP2α) was subsequently isolated from human HeLa cells, followed a few years later by the discovery of a second type II isoform, TOP2β, respectively [8,9,10,11,12].
Yeast TOP3 was the first type IA enzyme identified in eukaryotic organisms as a result of its role in homologous recombination [13, 14]. As with type II enzymes, vertebrates have two isoforms of TOP3: TOP3α and TOP3β [15, 16].
Since the original descriptions of topoisomerases, the function, structures, and mechanism of type I and type II classes of enzymes have been extensively studied [17,18,19,20,21]. DNA topoisomerase-targeted compounds, many of which are derived from natural products, have been shown to specifically bind to and stabilize the covalent topoisomerase–DNA reaction intermediates. Analogs of these compounds are FDA approved and/or are in development as antimicrobial and cancer chemotherapeutics [18, 22,23,24,25,26,27]. These chemotherapeutics enhance the stability of normally transient enzyme–DNA covalent complexes, which are then converted into lethal DNA lesions/breaks through interactions with replication and transcription machinery, or during chromosome segregation [19, 21, 23, 28,29,30,31,32,33]. For the purpose of this review, we will focus on chemotherapeutics that target eukaryotic topoisomerases. Human cells express six DNA topoisomerases representing three subfamilies: Type IA (TOP3α and TOP3β), type IB (TOP1 and TOP1mt), and type II (TOP2α and TOP2β) [17,18,19, 21].
Physiological function
DNA topoisomerases function to resolve topological problems that are intrinsic to the helical, duplex structure of DNA and are generated during DNA metabolism and gene expression. These enzymes catalyze the relaxation of DNA supercoils, and the decatenation and unknotting of DNA helices and strands. As such, DNA topoisomerases play essential roles in DNA replication, transcription, and repair, and in the condensation and segregation of chromosomes during mitosis [17,18,19].
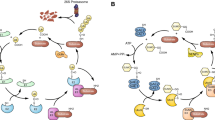
Eukaryotic topoisomerases are classified as type I or type II enzymes, based on the number of strands in duplex DNA that are transiently cleaved during the reaction cycle (one strand for type I and staggered double-strand breaks for type II). Categorization into IA, IB, and IIA subfamilies is based on differences in structure and mechanism (Table 1) [17,18,19, 34]. Type IA and type IB topoisomerases are largely monomeric enzymes that cleave one strand of duplex DNA. However, they do not share sequence or structural similarities, they form distinct phospho-tyrosyl linkages, and they exhibit different mechanisms of DNA supercoil relaxation (Fig. 1a, b). Type II topoisomerases induce a double-strand break, function as a dimer, and require ATP hydrolysis for the alterations in enzyme structure that accompany DNA strand passage (Fig. 1c). Recently, structural homologs of the type II archaeal topo VIB enzyme subunit have been identified in plants and animals and have been implicated in the formation of meiotic double-strand DNA breaks, after interaction with the SPO11 subunit [35]. While exhibiting structural and mechanistic similarities to type IIA enzymes, these topoisomerases are currently not therapeutic targets.

Three distinct catalytic mechanisms of topoisomerases. Each of the three types of DNA topoisomerases exhibits unique catalytic mechanism to alter DNA topology. a Type IA acts as a monomer and cleaves one strand forming a 5′-phospho-tyrosyl linkage within the protein–DNA complex. The opening of the cleaved strand allows the non-cleaved strand to pass through for DNA relaxation or (de)catenation. The ends of the cleaved strand religate to restore the phosphodiester backbone of the DNA and allowing the enzyme to dissociate from the DNA 5′-end. b Type IB also acts as a monomer and cleaves one strand of duplex DNA, but forms a transient 3′-phospho-tyrosyl bond. DNA relaxation occurs by the controlled rotation of the free 5′-end of the DNA around the non-cleaved strand. c Type IIA topoisomerases act as homodimers, with each subunit cleaving one strand and forms a 5′-phospho-tyrosyl linkage, resulting in a staggered double-strand break within the protein–DNA complex. DNA relaxation or (de)catenation occurs by passage of a double-stranded DNA molecule through the enzyme-linked double-strand break. Religation of the cleaved strands restores DNA integrity. See text for more details
To catalyze alterations in DNA topology, all DNA topoisomerases utilize successive transesterifications, where the active site tyrosine acts in the nucleophilic attack on the DNA phosphodiester backbone to form a transient phospho-tyrosyl linkage with the nicked DNA. This enzyme–DNA covalent complex is also referred to as a cleavable complex, as it may be irreversibly trapped with denaturing agents. Topological strain in the DNA is then released via topoisomerase-mediated strand passage for type IA and type II enzymes, or by a mechanism of enzyme-controlled strand rotation for type IB enzymes [19] (Fig. 1). Subsequent religation of the DNA strand is accomplished by a second transesterification reaction involving the nucleophilic attack of the free −OH group of the DNA end to resolve the phosphoryl-tyrosine bond. In these reactions, a water molecule has been posited to act as a general acid–base catalyst [34, 36].
Structure and function of DNA topoisomerase subfamilies
Type I topoisomerases
Eukaryotic type I topoisomerases can be divided into two subclasses; type IA includes topoisomerase III (TOP3α and TOP3β), and type IB consists of nuclear topoisomerase I (TOP1) and the mitochondrial isoform (TOP1mt) found only in vertebrates (Table 1). With the exception of the dimeric TOP1B from Trypanosoma and Leishmania parasites [37, 38], these enzymes function as monomers and consist of conserved structural elements. Sequence and mechanistic similarities of TOP1 and TOP1mt suggest that they share a common ancestral gene [39]. However, TOP1mt contains a much shorter N-terminal domain (51 residues versus 215 residues for nuclear TOP1), which contains a mitochondrial localization signal (MLS) and lacks the nuclear localization signal (NLS) found in TOP1. The core, linker, and C-terminal domains of these isoforms are sufficient for catalytic activity and show high degrees of identity (73, 57, and 75%, respectively). Both type IB isoforms are also the cellular target of the camptothecin (CPT) class of chemotherapeutics [4, 28, 40, 41]. In cells, TOP1mt is not affected by the parental compound CPT, but can be targeted by the topotecan analog, which is able to enter the mitochondrial matrix [42, 43]. However, it remains unclear what role targeting TOP1mt plays in the therapeutic response of cancer patients to toptotecan, or the prodrug CPT-11.
Mammalian cells express two isoforms of TOP3 (TOP3α and TOP3β). TOP3α can be found in the nucleus and mitochondria [44], and functions with the BLM helicase in a complex with RMI1 in the resolution of Holliday junctions and in replication fork recovery [45, 46]. TOP3β has also been shown to possess RNA topoisomerase activity on circular RNA (circRNA) substrates and its function in a ribonucleoprotein complex is required for normal neurodevelopment [47]. The TOP3 isoforms act to maintain genome stability and neurodevelopment and have not been the target of drug discovery efforts. Conversely, the prokaryotic type IA enzymes exhibit structural and mechanistic similarities to TOP3 and are potential targets for antibiotics to treat tuberculosis [26]. Thus, there may be complications associated with the chemotherapeutic targeting of tuberculosis in human populations where TOP3α and TOP3β are ubiquitously expressed.
The structures of type IA topoisomerases are generally described as a padlock, where domains I, III, and IV form the bottom of the structure (aka the lock), and domain II forms a loop that facilitates opening and closing of the nicked single strand of DNA [19, 48]. Mechanistically, type IA topoisomerases generate a transient 5′-phospho-tyrosyl bond that covalently links the enzyme with the 5′-end of the nicked single strand of duplex DNA (Fig. 1a), in a reaction that is Mg2+ dependent, but ATP independent. Type IA enzymes selectively catalyze the relaxation of negative supercoils in duplex DNA, where the formation of the covalent enzyme–DNA complex is facilitated by the propensity for negatively supercoiled duplex DNA to locally unwind. Consequently, type IA enzymes are unable to relax overwound, or positively supercoiled DNA. These enzymes also act as decatenases to disentangle precatenanes formed during DNA replication, and can catalyze the knotting/unknotting of single-stranded DNA. In all cases, these enzymes act by a strand transfer mechanism whereby the nonscissile strand is passed through the protein-linked nick in the opposite strand. Subsequent nucleophilic attack by the free 3′-hydroxyl end on the 5′-phospho-tyrosyl linkage results in religation of the DNA strand, allowing for dissociation of the enzyme from the DNA (Fig. 1a).
The type IB subfamily of topoisomerases does not share sequence, mechanistic, or structural homology with type IA or type II topoisomerases. Type IB enzymes are unique in the formation of covalent intermediates with a 3′-phospho-tyrosyl linkage, as opposed to the 5′-linkage observed with all other classes of topoisomerases (Fig. 1b). Unlike type IA enzymes, type IB topoisomerases are able to relax positive and negative supercoils, through a mechanism of DNA strand rotation that does not require Mg2+ [34]. In this case, topological stresses in the duplex DNA substrate, rather than the enzyme itself, dictates DNA strand unwinding or rewinding. However, as with other topoisomerases, the cleavage and religation of DNA strands does not require ATP.
The most widely studied type IB enzyme is human DNA topoisomerase I, for which several X-ray crystal structures have been resolved, all of which contain DNA substrates and, in many instances, drugs such as CPTs that target TOP1B [49, 50]. However, the N-terminally truncated TOPO70 enzyme used for these structural determinations lacks residues 1–174 of human TOP1B and, in these TOPO70 structures, only residues 203–765 have been resolved (Fig. 2a). Nevertheless, TOPO70 is able to bind DNA, is catalytically active, and is comparable to TOP1mt in size, reaction mechanism, and presumably tertiary structure [34]. TOPO70 consists of a core domain (core-subdomain I, II, & III), an extended pair of alpha helices that form the linker domain, and a C-terminal domain that contains the active site tyrosine [49, 50]. The core domain forms a “C”-type protein clamp, where domains I and II form the “cap,” while domain III forms the base of the “C.” The coiled-coil structure of the linker domain runs like a guided ruler away from the “C”-clamp and positions the active site tyrosine of the C-terminal domain within the enzyme active site formed by the core domain of the protein clamp. The “C”-clamp cradles the DNA, while in co-crystal structures that contain CPT or other TOP1 poison, the linker domain is positioned at an oblique angle to the double-stranded helical DNA that extends from the clamp, 3′ to the site of cleavage in the nicked DNA strand (Fig. 2) [49, 50]. In contrast, the linker domain is not resolved in covalent TOPO70–DNA complexes that lack drug [49]. In conjunction with reconstitution studies reported by Champoux and colleagues, these data suggest that the physical linkage and the flexibility of the linker domain impact the intrinsic sensitivity of TOP1B to CPT [51].

Type IB DNA topoisomerase. a Human TOP1B (TOPO70) crystal structure with DNA and topotecan (TPT). Surface representation of the N-terminally truncated TOPO70 (lacking the first 174 residues) in orange with DNA (sticks) and TPT (blue balls), PDB# 1K4T [100]. All structure figures were generated using MacPyMol 0.99rc6 (DeLano Scientific, San Carlos, CA). b Schematic representation of eukaryotic DNA topoisomerase I (TOP1B) catalytic cycle where the covalent enzyme–DNA intermediate constitutes the sole cellular target of camptothecin (CPT). Topotecan and irinotecan as FDA-approved CPT analogs. TOP1B non-covalently circumscribes duplex DNA. The active site tyrosine cleaves one strand forming a 3′-phospho-tyrosyl bond, which covalently links the enzyme to the 3′-end of DNA. After DNA relaxation by a controlled rotation mechanism, the DNA ends religate and TOP1B may dissociate from the DNA. CPT intercalates into the cleavage site preventing religation of the DNA ends. The reversible drug-stabilized enzyme–DNA complex can be converted into lethal DNA lesions in collisions with processive replication or transcription machinery. See text for more details
In terms of reaction mechanism, type IB enzymes first bind non-covalently to double-stranded DNA, where the “C”-clamp completely circumscribes the DNA, and the active site tyrosine is poised for nucleophilic attack of the phosphodiester backbone bond of the scissile DNA strand. Upon DNA strand cleavage and concomitant linkage of the enzyme to the 3′- phosphoryl DNA end, the free 5′-hydroxyl DNA end rotates within the protein clamp, around the non-cleaved strand, to alleviate DNA strand over- or underwinding (positive or negative supercoils, respectively). The covalent complex is resolved by a second transesterification reaction, involving the nucleophilic attack by the 5′-hydroxyl DNA end to reform the phosphodiester bond in the DNA. The enzyme subsequently dissociates from the non-covalent enzyme DNA complex or initiates a new DNA cleavage–religation cycle at the same site (Figs. 1b, 2b).
Type II topoisomerases
Eukaryotic type II topoisomerases function as homodimers and transiently cut both strands of a DNA duplex. Then, through a strand passage mechanism (Fig. 1c), these enzymes catalyze the relaxation of positively and negatively supercoiled DNA, or the decatenation/unknotting of entangled double-stranded DNA molecules. Mammalian cells express two type IIA isoforms (TOP2α and TOP2β). TOP2α expression is cycle dependent, increasing in S phase and peaking during G2/M phase, to assist in the untangling of DNA duplexes during replication. This enzyme also catalyzes the relaxation of positive supercoiled DNA more efficiently than negative supercoils and decatenates mitotic chromosomes during cell division. In contrast, TOP2β is constitutively expressed in replicating and quiescent cells. TOP2β plays a critical role during neuronal cell development and has been implicated in the regulation of gene transcription and DNA recombination, yet acts equally efficiently in the relaxation of positively and negatively supercoiled DNA [52,53,54,55].
Human TOP2α and TOP2β share considerable mechanistic, structural, and sequence identity with each other and the related type II enzyme expressed in the yeast Saccharomyces cerevisiae, being 68% identical in amino acid sequence to each other and 40% identical to yeast TOP2. Numerous structures of the C-terminal catalytic core and N-terminal ATPase domains of eukaryotic DNA type II enzymes have been separately resolved, as well as a near-complete structure of yeast TOP2 (Fig. 3) [56,57,58,59]. Given the overall conservation of the eukaryotic and bacterial type IIA family of enzymes, the compilation of these structures provides detailed mechanistic insights into type II enzyme catalysis and drug targeting [21, 57, 59, 60].

Crystal structures of eukaryotic type IIA topoisomerases. a Near-complete structure of yeast TOP2 PBD# 4GFH [59]. TOP2 protein surface is shown in blue/cyan with DNA (sticks) and the non-hydrolysable ATP analog AMPNP (green balls). b C-terminal catalytic core surface structure of human TOP2β shown in pale blue/pale cyan, DNA (sticks), and etoposide -VP16- (gray balls) from PDB# 3QX3 [57]
A series of dimer interfaces form three gates in the type IIA homodimer through which a DNA duplex is passed (Fig. 4). A series of coordinated, ATP-dependent conformational changes in enzyme promoter structure allow for gate opening and closing, to pass one DNA duplex (the T-segment) through a staggered double-stranded break in a second segment of duplex DNA (G-segment). The N-gate is formed by the N-terminal ATPase domain of each promoter, which consists of the GHKL (Gyrase, Heat-shock protein 90, histidine Kinase, and mutL) domain and a structure-relaying transducer domain [54, 59, 61]. These domains are linked to the catalytic core of the enzyme, which forms the DNA-gate and comprises a metal-chelating TOPRIM (topoisomerase/primase) domain, a winged-helix domain (WHD) containing the active site tyrosine, and a tower domain. The third dimerization interface, the C-gate, consists of a coiled-coil domain that links the structural elements of the DNA-gate to the largely disordered C-terminal domain (Fig. 4) [21, 56, 57, 60,61,62].

DNA Topoisomerase II catalytic cycle. Topoisomerase II binds two duplex DNAs. First, the Gated or G-segment DNA stands enters the open N-gate formed by the dimeric ATPase domains. The Transported or T-segment DNA (black) then enters via the N-gate. The T-segment DNA may be from the same molecule as the G-segment DNA (during DNA relaxation or (un)knotting), or from another DNA molecule [(de)catenation]. Subsequently, two ATP molecules bind to the ATPase domains, stimulating domain dimerization to close the N-gate. Upon hydrolysis of one ATP molecule, G-segment DNA strands are cleaved and covalently linked to one subunit via a 5′-phospho-tyrosyl bond. The T-segment DNA is then transported through the cleaved G-segment (G-gate). The strands of the G-segment are religated and the remaining ATP is hydrolyzed. Upon dissociation of the two ADP molecules, the T-segment is released through the C-terminal “C-gate.” Closing of the C-gate is followed by separation of the ATPase domains to allow the G-segment to leave the enzyme complex. Doxorubicin (Dox) or adriamycin can inhibit the catalytic cycle at two different steps. Dox blocks the religation of the cleaved G-segment, which is also the mechanism of action for etoposide (VP16). However, Dox can also interfere with DNA binding at the start of the catalytic cycle, preventing TOP2-mediated DNA relaxation or (de)catenation. ICRF-187 prevents the hydrolysis of the second ATP molecule, preventing separation of the ATPase domains or opening of the N-gate. See text for more details
As shown in Fig. 4 and Table 1, type IIA topoisomerase catalysis is dependent on ATP and Mg2+ and involves the preferential binding of duplex DNA crossovers (the G- and T-segments) [63]. ATP binding induces dimerization of the N-gate, to trap the G- and T-segments within the closed protein clamp. Subsequently, the active site tyrosines in each promoter cleave opposite DNA strands to form staggered 5′-phospho-tyrosyl bonds. The broken G-segment is then pulled apart to allow the T-segment to be transported into the bottom C-gate. Hydrolysis of one ATP molecule promotes T-segment passage through the DNA-gate. The G-segment is then religated and the T-segment is released through the C-gate. Hydrolysis of the second ATP resets the open N-gate and the G-segment is released. Recent structural studies suggest that limited conformational changes in the WHDs may couple DNA cleavage/religation with the allosteric control of C-gate opening [56].
Topoisomerases as drug targets
The intertwining of DNA strands and helices, produced during essential cellular processes of replication, recombination, transcription, and chromosome segregation, must be resolved in order to maintain cell viability and genome stability. DNA topoisomerases provide an important solution for resolving such topological problems. However, they do so through the formation of a covalent enzyme–DNA reaction intermediate, which is in itself a potentially toxic lesion. Indeed, targeting of topoisomerase–DNA complexes has been widely exploited in the identification and development of antibacterial and anticancer chemotherapeutics [17, 18, 23, 26]. The majority of these agents are termed “poisons” to indicate a mechanism of covalent enzyme–DNA complex stabilization (Figs. 2a, 3b), versus “inhibitor,” which would signify the lack of DNA binding or cleavage by the enzyme. A critical aspect of these differences in the mechanism of drug action is that in an isogenic cell system, increased expression of the enzyme would increase the cytotoxic activity of a “poison” through increased production of drug-stabilized topoisomerase–DNA adducts. In contrast, elevated levels of enzyme would confer resistance to “inhibitors.” FDA-approved topoisomerase IB (Fig. 5) and type IIA (Fig. 6) poisons, such as topotecan and etoposide, respectively, are analogs of naturally occurring poisons, camptothecin (Fig. 5a, b) and podophyllotoxin (Fig. 6a), respectively. (For drug information, see: https://www.cancer.gov/publications/dictionaries/cancer-drug).

Structures of DNA topoisomerase IB inhibitors. Shown are the structures of camptothecin or analogs that are currently FDA approved (a), or in clinical trials (b). c Non-camptothecin agents currently in clinical trials

Structures of DNA topoisomerase II inhibitors. Shown are agents that are currently FDA approved (italic), or in clinical trials (normal font): a podophyllotoxin analogs, b anthracyclines analogs, and c other agents
In addition, many of the topoisomerase-targeting agents act as interfacial inhibitors (poisons) [64]. Interfacial inhibitors are different from competitive (orthosteric) inhibitors and non-competitive (allosteric) inhibitors since they interact at the interface between two or more molecules. TOP1B-targeting CPTs are a great example of interfacial inhibitors; these compounds interact with the protein–DNA interface, effectively acting as additional nucleotides to prevent DNA religation (reviewed in [65]). Moreover, elegant work in yeast showed that the TOP1B inhibitors act as poisons rather than catalytic inhibitors [66, 67].
TOP1B poisons
Camptothecin was isolated from the chinese tree Camptotheca acuminata and identified in a natural products screen for anti-leukemic activity [41]. Although active in cancer clinical trials, low solubility and adverse drug reactions limited clinical development [68, 69]. Subsequent development efforts yielded several water-soluble CPT analogs, with improved toxicity profiles. Two of these—topotecan and the prodrug irinotecan—are FDA approved for the treatment of ovarian and lung cancer, and colorectal carcinoma, respectively [22, 23, 28]. All CPT analogs reversibly stabilize the TOP1B–DNA complex. In this ternary drug–enzyme–DNA complex, CPT-induced misalignment of the 5′-hydroxyl group and the scissile tyrosine–DNA bond prevents DNA religation (Fig. 2). However, these protein-linked DNA lesions are not toxic in themselves. Rather, the collision of DNA replication forks with the ternary complexes, or the positively supercoiled DNA domains induced by these complexes, produces the irreversible DNA lesions and double-strand breaks that ultimately lead to cell death [33, 70, 71]. Thus, the cytotoxicity of these drugs is S phase dependent.
A common aspect of all CPT analogs is a planar pentacyclic structure, where the conjugated rings are labeled A–E. The lactone ring is highly susceptible to hydrolysis, and at physiological pH, ring E is in equilibrium between the closed lactone and open carboxylate forms (Fig. 5a). However, the lactone form is more lipophilic, which facilitates cellular uptake, and is required for productive interactions with the TOP1B–DNA complex. The addition of a methyl group to the E ring, to form a 7-member β-hydroxyacetone in homocamptothecin, slows ring opening and enhances potency [72]. Several homocamptothecins have been evaluated in clinical trials; however, irreversible ring opening and increased protein binding have limited further clinical application. Another E ring modification in the prodrug camptothecin-20(S)-O-propionate hydrate (CZ48) is the addition of propionate ester, which stabilizes the lactone ring (Fig. 5b). Subsequent cleavage by cellular esterases yields active CPT. CZ48 is currently in phase I clinical trials.
Extensive analog development has determined that modification of the C, D, and E rings reduces the cytotoxic activity of CPT [23, 70, 73, 74]. On the other hand, modifications to the A ring at positions 9, 10, and 11 and position 7 of the B ring have been shown to be well tolerated and provide improvements in pharmacologic properties and potency [23, 70, 73, 74]. The semisynthetic CPT derivatives irinotecan and topotecan received FDA approval in 1996 (Fig. 5a). Topotecan is a water-soluble derivative, with position 8 and 9 substituents, and is also approved for oral use. Bone marrow suppression, primarily neutropenia, is the dose-limiting toxicity. Irinotecan (CPT-11) is a prodrug with substituents at positions 7 and 10, which, when cleaved by serum or cellular carboxylesterases, yields the active metabolite SN-38 (Fig. 5a). Dose-limiting toxicities for irinotecan include neutropenia and diarrhea [75]. The latter results from the elimination of SN-38 and its glucuronidated form in the bile and feces. The activity of bacterially expressed β-glucuronidase in the gut generates active SN-38, which in turn induces epithelial cell damage associated with diarrhea. Prophylactic treatment with antibiotics can mitigate this dose-limiting toxicity [76].
Numerous A- and B-ring substituents have been evaluated in preclinical and clinical trials, including 9-amino CPT and 9-nitro CPT analogs, and hexacyclic derivatives with rings at positions 7/8 or 9/10. The addition of alkyl groups at position 7 was found to increase the cytotoxic activity of CPT. Among these analogs, the improved water solubility of belotecan (CKD-602) results from a basic nitrogen incorporated into an alkyl chain at position 7 (Fig. 5a). This analog is currently in clinical trials and is approved in South Korea for the treatment of small cell lung cancer and ovarian cancer. Among the most potent CPT analogs is gimatecan (ST1481), an oxyiminomethyl derivative (position 7) that is not a substrate for ATP-binding cassette transporters and, as a consequence, is orally bioavailable [77, 78]. Limited phase I–II trials were completed in 2015, while the results from a Chinese phase II clinical trial (2016) have yet to be reported. Silatecan contains an alkylsilyl group at position 7 and an −OH group at position 10, which reduces protein binding and allows the drug to cross the blood–brain barrier (Fig. 5b) [79]. This CPT analog, also known as DB-67 or AR-67, is currently in phase II clinical development. Another 7-silyl CPT, karenitecin (BNP1350), has completed clinical trial evaluations and is not currently in active trials [73, 80, 81].
A novel class of non-camptothecin TOP1B “poisons” is indenoisoquinolines (Fig. 5c). These synthetic molecules exhibit improved chemical stability and longer stabilization of the covalent TOP1B–DNA intermediate than that observed with CPTs [82]. Of a series of over 400 analogs, two derivatives, indotecan (LMP400) and indimitecan (LMP776), are currently being evaluated in phase I clinical trials. LMP400 shows no significant gastrointestinal problems, as are observed with CPT-11, but induce some myelosuppression [83]. Moreover, indenoisoquinolines can overcome multidrug resistance, as they are not substrates for the drug efflux transporters, which limits the oral bioavailability of CPTs [84].
Type IIA topoisomerase poisons
In the mid-1960s, two podophyllotoxin analogs, the epipodophyllotoxin etoposide and the closely related teniposide (Fig. 6a), were identified from a series of synthetically derived compounds exhibiting potent antineoplastic activity [85]. Podophyllotoxin is derived from podophyllin, an alcohol extract of dried roots and rhizomes of the plant Podophyllum peltatum, also known as mandrake [86]. In 1983, etoposide received FDA approval as a cancer chemotherapeutic, although its mechanism of action was not fully understood [85]. The following year, Leroy Liu and colleagues reported etoposide and teniposide “poisoning” of DNA topoisomerase II [87]. Similar to CPT poisoning of TOP1B, etoposide stabilizes the topoisomerase II–DNA reaction intermediate (Fig. 3b), preventing religation of the DNA strands, which causes cell death (Fig. 4). Teniposide was approved by the FDA in 1992.
Etoposide comprises three chemical moieties: a polycyclic core (A–D rings), a glycosidic group (attached to position 4 position of the C ring), and a pendant ring (E ring) [25, 85]. The pendant ring is crucial for the activity of etoposide. Modification of the C-4 glycosidic group can also affect drug activity; however, it does not aid etoposide interactions with topoisomerase II [85]. Teniposide differs from etoposide only by a thienyl glycoside group in place of a methyl (Fig. 6a) [24].
Based on the activity of these epipodophyllotoxins, considerable efforts were made to develop more effective analogs. Structure–activity relationship (SAR) studies of A and B rings resulted in decreased potency relative to etoposide, as did an extensive SAR campaign of E-ring substituents. However, the addition of an E-ring phosphate ester in the prodrug etopophos improved the water solubility of etoposide and allowed for oral bioavailability. Etopophos is readily converted to the parent compound and is currently in the clinic. Another E-ring analog, CAP7.1, a prodrug that is converted to etoposide by carboxylesterases, is in clinical trials in Germany. C-ring substituents, particularly at position C7, have yielded improved activity profiles, with several analogs [such as TOP-53 (Fig. 6a)] in preclinical development [88].
Other classes of FDA-approved topoisomerase II “poisons” include the anthracycline antibiotics, daunorubicin and its hydroxylated congener doxorubicin (Adriamycin), which are derived from Streptomyces, and their analogs idarubicin and epirubicin, respectively (Fig. 6b). These drugs intercalate into double-stranded DNA and interfere with the cleavage–ligation cycle of DNA topoisomerase II. Doxorubicin is perhaps one of the most active cancer therapeutics developed (Fig. 4). However, a serious side effect is cardiomyopathy, which can lead to congestive heart failure. Initially attributed to doxorubicin generation of oxygen free radicals, recent studies suggest that cardiac toxicity results from drug poisoning of the TOP2β isoform of topoisomerase II [89]. Other factors have also been implicated in doxorubicin-induced cardiotoxicity. TOP1mt deficiency was recently linked with enhanced doxorubicin-induced cardiomyocyte toxicity [90]. TOP1mt is a critical factor in normal mtDNA homeostasis and may assist in repair of damaged mtDNA [91]. Indeed, Khiati and co-workers observed numerous genomic variants for TOP1mt, within the general population, which have the potential to impair TOP1mt [90].
As with other topoisomerase-targeted drugs, analog development has been pursued to improve the safety profile and efficacy of anthracyclines. One such derivative of doxorubicin, amrubicin, has received approval in Japan; however, it has only been awarded orphan drug status by the FDA (Fig. 6b). Another analog, esorubicin, exhibited reduced cardiotoxicity, but was also associated with increased myelosuppression. Several approaches have been shown to be more effective in reducing the risk of cardiotoxicity, without affecting tumor response to doxorubicin, including the use of the bisdioxopiperazine topoisomerase II catalytic inhibitor dexrazoxane [ICRF-187] (Figs. 4, 6c). Another approach is the development of nanoparticle formulations that limit drug delivery to the heart, such as liposomal PEGylated doxorubicin (DOXIL). Other formulations in clinical development include glutathione PEGylated liposomal doxorubicin (2B3-101) for selective delivery to the brain, HER2 antibody liposomal doxorubicin (MM-302) for targeting of HER2-expressing tumors, and a co-polymer of N-(2-Hydroxypropyl) methacrylamide (HPMA) and doxorubicin (FCE2806) with no cardiotoxicity. Aldoxorubicin is a 6-maleimidocaproyl hydrazone derivative prodrug undergoing clinical evaluation, where doxorubicin is released in the tumor microenvironment following cleavage of the acid-labile hydrazine linker.
A third class of FDA-approved topoisomerase II poisons is mitoxantrone (Fig. 6c). It is a DNA-reactive agent that intercalates into DNA through hydrogen bonding, and causes crosslinks and strand breaks [92, 93]. It binds to topoisomerase II forming a cleavable complex that will induce DNA strand breaks and successively apoptosis [94].
In contrast to poisons, topoisomerase II catalytic inhibitors do not stabilize the covalent enzyme–DNA intermediate at pharmacologically relevant concentrations. In addition to the bisdioxopiperazine dexrazoxane (ICRF-187) mentioned above in the discussion of doxorubicin, other examples of structurally unrelated classes of inhibitors include the bisdioxopiperazine ICRF-193, the anthracycline aclarubicin, merbarone, and the coumarin drug novobiocin [95]. Of these, dexrazoxane and aclarubicin are currently in clinical development (Fig. 6).
Clinical challenges
As discussed above, numerous TOP1B- and topoisomerase II-targeted agents and analogs have been developed and assessed in preclinical and clinical studies. Clinicaltrials.gov currently lists over 900 active clinical trials of topoisomerase-targeted agents, ~800 of which are cancer trials (for an example of open clinical trials, see Table 2). Of these, ~430 are in the US, 57 in Canada, 200 in the EU, and 160 in China. However, only a handful of these trials involve novel drugs as single agents. Topoisomerases have proven critical cancer targets, with a significant number of FDA-approved agents as effective cytotoxic therapies for the treatment of solid and liquid malignancies in adult and pediatric cancer patient populations. Nevertheless, current clinical challenges are focused primarily on two critical issues: (1) combining these effective agents with other cytotoxic and molecularly targeted agents, and (2) new formulations to improve the pharmacologic properties of these drugs and overcome the limitations of scheduling.
Etoposide, doxorubicin, and irinotecan have been incorporated in multiple chemotherapeutic regimens for the treatment of a wide range of solid malignancies. In addition to clinical investigations of new combinations of cytotoxic drug regimens and multi-modality regimens with radiation, current clinical trials include combining these topoisomerase poisons with molecularly targeted therapeutics, such as poly(ADP-ribose) polymerase (PARP) inhibitors, signaling inhibitors (rapamycin analogs), immune checkpoint modulators, and DNA damage checkpoint inhibitors (Table 2). Obviously, the challenge here is to develop robust preclinical models, such as patient-derived xenografts (PDX), and gene expression/editing technology-based screening (RNAi and CRISPR/Cas9) to define the synthetic lethal interactions predictive of pathway interactions that may be exploited with novel drug combinations. An interesting example is preclinical studies demonstrating the increased bioavailability of irinotecan and topotecan, induced in mice by HER tyrosine kinase inhibitor targeting of the ABCG2 drug transporter [96, 97].
In the context of topoisomerase catalysis, the existence of specific tyrosyl-DNA phosphodiesterases, TDP1 and TDP2, allows the development of targeted synthetic lethal drug combinations. TDP1 and TDP2 are able to hydrolyze the covalent phospho-tyrosyl linkage between the topoisomerase active site tyrosine and the phosphoryl end of the DNA. TDP1 preferentially resolves a 3′-phospho-tyrosyl link to the 3′-DNA end, while TDP2 prefers a 5′-phospho-tyrosyl bond which attaches TOP2 to the 5′-DNA end (review in [98]). Consequently, synthetic lethality would be predicted to result from the inhibition of TDP1 or TDP2 activity in combination with a TOP1B or a TOP2 poison, respectively. However, this concept will have to wait for future validation, as no TDP (1 or 2) inhibitors are currently available for clinical evaluation.
As described above for doxorubicin, the clinical development of novel nanoparticle delivery systems is also being exploited to avoid non-malignant tissue toxicity, improve tumor targeting, and address issues of drug stability, oral dosing, and scheduling. In the case of CPT targeting of TOP1B, the S-phase dependent cytotoxic activity of these agents presents challenges in the optimal daily dosing of drugs to maximize therapeutic efficacy (Table 2). Efforts to increase the half-life of CPT analogs could provide for prolonged exposure of S-phase populations of tumor cells, without repeated dosing. Along these lines, the FDA has approved a long-lived, PEGylated nanoliposomal formulation of irinotecan (Onivyde) for use in combination with fluorouracil and leucovorin, for the treatment of metastatic adenocarcinoma of the pancreas after disease progression. Santi and colleagues at Prolynx recently reported the development of PEG conjugates of SN-38, where β-elimination of phenol ether linkages releases SN-38 with a predictable half-life [99]. Irinotecan or its active metabolite SN-38 has also either been PEGylated (e.g., NKTR102) or attached to nanoparticles (e.g., NK012) to enhance pharmacodynamics and tumor delivery. Moreover, a nano-formulation of CPT in CRLX101, which is water soluble, is currently in clinical trials for the treatment of small cell lung cancer in combination with the PARP inhibitor Olaparib. For current updates of clinical trial of these modified TOP1 and TOP2 inhibitors, see https://clinicaltrials.gov/. The application of these and other novel technologies promises to overcome some of the obstacles in drug dosing and delivery that have hampered the clinical application of what are otherwise extremely active cancer therapeutics.
References
Wang JC (1971) Interaction between DNA and an Escherichia coli protein omega. J Mol Biol 55(3):523–533
Champoux JJ, Dulbecco R (1972) An activity from mammalian cells that untwists superhelical DNA–a possible swivel for DNA replication (polyoma-ethidium bromide-mouse-embryo cells-dye binding assay). Proc Natl Acad Sci USA 69(1):143–146
Champoux JJ (1981) DNA is linked to the rat liver DNA nicking-closing enzyme by a phosphodiester bond to tyrosine. J Biol Chem 256(10):4805–4809
Zhang H, Barcelo JM, Lee B, Kohlhagen G, Zimonjic DB, Popescu NC, Pommier Y (2001) Human mitochondrial topoisomerase I. Proc Natl Acad Sci USA 98(19):10608–10613. doi:10.1073/pnas.191321998
Gellert M, Mizuuchi K, O’Dea MH, Nash HA (1976) DNA gyrase: an enzyme that introduces superhelical turns into DNA. Proc Natl Acad Sci USA 73(11):3872–3876
Mizuuchi K, Fisher LM, O’Dea MH, Gellert M (1980) DNA gyrase action involves the introduction of transient double-strand breaks into DNA. Proc Natl Acad Sci USA 77(4):1847–1851
Liu LF, Liu CC, Alberts BM (1980) Type II DNA topoisomerases: enzymes that can unknot a topologically knotted DNA molecule via a reversible double-strand break. Cell 19(3):697–707
Austin CA, Fisher LM (1990) Isolation and characterization of a human cDNA clone encoding a novel DNA topoisomerase II homologue from HeLa cells. FEBS Lett 266(1–2):115–117
Baldi MI, Benedetti P, Mattoccia E, Tocchini-Valentini GP (1980) In vitro catenation and decatenation of DNA and a novel eucaryotic ATP-dependent topoisomerase. Cell 20(2):461–467
Chung TD, Drake FH, Tan KB, Per SR, Crooke ST, Mirabelli CK (1989) Characterization and immunological identification of cDNA clones encoding two human DNA topoisomerase II isozymes. Proc Natl Acad Sci USA 86(23):9431–9435
Drake FH, Zimmerman JP, McCabe FL, Bartus HF, Per SR, Sullivan DM, Ross WE, Mattern MR, Johnson RK, Crooke ST et al (1987) Purification of topoisomerase II from amsacrine-resistant P388 leukemia cells. Evidence for two forms of the enzyme. J Biol Chem 262(34):16739–16747
Miller KG, Liu LF, Englund PT (1981) A homogeneous type II DNA topoisomerase from HeLa cell nuclei. J Biol Chem 256(17):9334–9339
Kim RA, Wang JC (1992) Identification of the yeast TOP3 gene product as a single strand-specific DNA topoisomerase. J Biol Chem 267(24):17178–17185
Wallis JW, Chrebet G, Brodsky G, Rolfe M, Rothstein R (1989) A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell 58(2):409–419
Hanai R, Caron PR, Wang JC (1996) Human TOP3: a single-copy gene encoding DNA topoisomerase III. Proc Natl Acad Sci USA 93(8):3653–3657
Seki T, Seki M, Onodera R, Katada T, Enomoto T (1998) Cloning of cDNA encoding a novel mouse DNA topoisomerase III (Topo IIIbeta) possessing negatively supercoiled DNA relaxing activity, whose message is highly expressed in the testis. J Biol Chem 273(44):28553–28556
Wang JC (2002) Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 3(6):430–440. doi:10.1038/nrm831
Pommier Y, Sun Y, Huang SN, Nitiss JL (2016) Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol 17(11):703–721. doi:10.1038/nrm.2016.111
Vos SM, Tretter EM, Schmidt BH, Berger JM (2011) All tangled up: how cells direct, manage and exploit topoisomerase function. Nat Rev Mol Cell Biol 12(12):827–841. doi:10.1038/nrm3228
Leppard JB, Champoux JJ (2005) Human DNA topoisomerase I: relaxation, roles, and damage control. Chromosoma 114(2):75–85. doi:10.1007/s00412-005-0345-5
Chen SH, Chan NL, Hsieh TS (2013) New mechanistic and functional insights into DNA topoisomerases. Annu Rev Biochem 82:139–170. doi:10.1146/annurev-biochem-061809-100002
Pommier Y (2006) Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 6(10):789–802. doi:10.1038/nrc1977
Pommier Y (2013) Drugging topoisomerases: lessons and challenges. ACS Chem Biol 8(1):82–95. doi:10.1021/cb300648v
Hande KR (1998) Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer 34(10):1514–1521
Nitiss JL (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9(5):338–350. doi:10.1038/nrc2607
Tse-Dinh YC (2016) Targeting bacterial topoisomerases: how to counter mechanisms of resistance. Future Med Chem 8(10):1085–1100. doi:10.4155/fmc-2016-0042
Aldred KJ, Kerns RJ, Osheroff N (2014) Mechanism of quinolone action and resistance. Biochemistry 53(10):1565–1574. doi:10.1021/bi5000564
Hsiang YH, Hertzberg R, Hecht S, Liu LF (1985) Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 260(27):14873–14878
Li TK, Liu LF (2001) Tumor cell death induced by topoisomerase-targeting drugs. Annu Rev Pharmacol Toxicol 41:53–77. doi:10.1146/annurev.pharmtox.41.1.53
Pommier Y, Schwartz RE, Kohn KW, Zwelling LA (1984) Formation and rejoining of deoxyribonucleic acid double-strand breaks induced in isolated cell nuclei by antineoplastic intercalating agents. Biochemistry 23(14):3194–3201
Tewey KM, Chen GL, Nelson EM, Liu LF (1984) Intercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J Biol Chem 259(14):9182–9187
Zwelling LA, Michaels S, Erickson LC, Ungerleider RS, Nichols M, Kohn KW (1981) Protein-associated deoxyribonucleic acid strand breaks in L1210 cells treated with the deoxyribonucleic acid intercalating agents 4′-(9-acridinylamino) methanesulfon-m-anisidide and adriamycin. Biochemistry 20(23):6553–6563
Hsiang YH, Lihou MG, Liu LF (1989) Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res 49(18):5077–5082
Champoux JJ (2001) DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 70:369–413. doi:10.1146/annurev.biochem.70.1.369
Robert T, Vrielynck N, Mezard C, de Massy B, Grelon M (2016) A new light on the meiotic DSB catalytic complex. Semin Cell Dev Biol 54:165–176. doi:10.1016/j.semcdb.2016.02.025
Redinbo MR, Champoux JJ, Hol WG (2000) Novel insights into catalytic mechanism from a crystal structure of human topoisomerase I in complex with DNA. Biochemistry 39(23):6832–6840
Villa H, Otero Marcos AR, Reguera RM, Balana-Fouce R, Garcia-Estrada C, Perez-Pertejo Y, Tekwani BL, Myler PJ, Stuart KD, Bjornsti MA, Ordonez D (2003) A novel active DNA topoisomerase I in Leishmania donovani. J Biol Chem 278(6):3521–3526. doi:10.1074/jbc.M203991200
Vlachakis D, Pavlopoulou A, Roubelakis MG, Feidakis C, Anagnou NP, Kossida S (2014) 3D molecular modeling and evolutionary study of the Trypanosoma brucei DNA Topoisomerase IB, as a new emerging pharmacological target. Genomics 103(1):107–113. doi:10.1016/j.ygeno.2013.11.008
Zhang H, Meng LH, Zimonjic DB, Popescu NC, Pommier Y (2004) Thirteen-exon-motif signature for vertebrate nuclear and mitochondrial type IB topoisomerases. Nucleic Acids Res 32(7):2087–2092. doi:10.1093/nar/gkh525
Adams DJ, Morgan LR (2011) Tumor physiology and charge dynamics of anticancer drugs: implications for camptothecin-based drug development. Curr Med Chem 18(9):1367–1372
Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA (1966) Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from camptotheca acuminata1,2. J Am Chem Soc 88(16):3888–3890. doi:10.1021/ja00968a057
Croce AC, Bottiroli G, Supino R, Favini E, Zuco V, Zunino F (2004) Subcellular localization of the camptothecin analogues, topotecan and gimatecan. Biochem Pharmacol 67(6):1035–1045. doi:10.1016/j.bcp.2003.10.034
de la Loza MC, Wellinger RE (2009) A novel approach for organelle-specific DNA damage targeting reveals different susceptibility of mitochondrial DNA to the anticancer drugs camptothecin and topotecan. Nucleic Acids Res 37(4):e26. doi:10.1093/nar/gkn1087
Wang Y, Lyu YL, Wang JC (2002) Dual localization of human DNA topoisomerase IIIalpha to mitochondria and nucleus. Proc Natl Acad Sci USA 99(19):12114–12119. doi:10.1073/pnas.192449499
Swuec P, Costa A (2014) Molecular mechanism of double Holliday junction dissolution. Cell Biosci 4:36. doi:10.1186/2045-3701-4-36
Chaudhury I, Sareen A, Raghunandan M, Sobeck A (2013) FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res 41(13):6444–6459. doi:10.1093/nar/gkt348
Nott A, Tsai LH (2013) The Top3beta way to untangle RNA. Nat Neurosci 16(9):1163–1164. doi:10.1038/nn.3506
Champoux JJ (2002) A first view of the structure of a type IA topoisomerase with bound DNA. Trends Pharmacol Sci 23(5):199–201
Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WG (1998) Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 279(5356):1504–1513
Stewart L, Redinbo MR, Qiu X, Hol WG, Champoux JJ (1998) A model for the mechanism of human topoisomerase I. Science 279(5356):1534–1541
Stewart L, Ireton GC, Champoux JJ (1997) Reconstitution of human topoisomerase I by fragment complementation. J Mol Biol 269(3):355–372. doi:10.1006/jmbi.1997.1056
Fortune JM, Osheroff N (2000) Topoisomerase II as a target for anticancer drugs: when enzymes stop being nice. Prog Nucleic Acid Res Mol Biol 64:221–253
McClendon AK, Osheroff N (2007) DNA topoisomerase II, genotoxicity, and cancer. Mutat Res 623(1–2):83–97. doi:10.1016/j.mrfmmm.2007.06.009
Berger JM, Gamblin SJ, Harrison SC, Wang JC (1996) Structure and mechanism of DNA topoisomerase II. Nature 379(6562):225–232. doi:10.1038/379225a0
Wang JC (1998) Moving one DNA double helix through another by a type II DNA topoisomerase: the story of a simple molecular machine. Q Rev Biophys 31(2):107–144
Wendorff TJ, Schmidt BH, Heslop P, Austin CA, Berger JM (2012) The structure of DNA-bound human topoisomerase II alpha: conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J Mol Biol 424(3–4):109–124. doi:10.1016/j.jmb.2012.07.014
Wu CC, Li TK, Farh L, Lin LY, Lin TS, Yu YJ, Yen TJ, Chiang CW, Chan NL (2011) Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 333(6041):459–462. doi:10.1126/science.1204117
Wei H, Ruthenburg AJ, Bechis SK, Verdine GL (2005) Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase. J Biol Chem 280(44):37041–37047. doi:10.1074/jbc.M506520200
Schmidt BH, Osheroff N, Berger JM (2012) Structure of a topoisomerase II-DNA-nucleotide complex reveals a new control mechanism for ATPase activity. Nat Struct Mol Biol 19(11):1147–1154. doi:10.1038/nsmb.2388
Schoeffler AJ, Berger JM (2005) Recent advances in understanding structure-function relationships in the type II topoisomerase mechanism. Biochem Soc Trans 33(Pt 6):1465–1470. doi:10.1042/BST20051465
Roca J, Wang JC (1994) DNA transport by a type II DNA topoisomerase: evidence in favor of a two-gate mechanism. Cell 77(4):609–616
Aravind L, Leipe DD, Koonin EV (1998) Toprim–a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res 26(18):4205–4213
Schmidt BH, Burgin AB, Deweese JE, Osheroff N, Berger JM (2010) A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Nature 465(7298):641–644. doi:10.1038/nature08974
Pommier Y, Marchand C (2005) Interfacial inhibitors of protein-nucleic acid interactions. Curr Med Chem Anticancer Agents 5(4):421–429
Pommier Y, Kiselev E, Marchand C (2015) Interfacial inhibitors. Bioorg Med Chem Lett 25(18):3961–3965. doi:10.1016/j.bmcl.2015.07.032
Bjornsti MA, Benedetti P, Viglianti GA, Wang JC (1989) Expression of human DNA topoisomerase I in yeast cells lacking yeast DNA topoisomerase I: restoration of sensitivity of the cells to the antitumor drug camptothecin. Cancer Res 49(22):6318–6323
Nitiss J, Wang JC (1988) DNA topoisomerase-targeting antitumor drugs can be studied in yeast. Proc Natl Acad Sci USA 85(20):7501–7505
Wall ME, Wani MC (1995) Camptothecin and taxol: discovery to clinic–thirteenth Bruce F. Cain Memorial Award Lecture. Cancer Res 55(4):753–760
Wall ME, Wani MC (1996) Camptothecin and taxol: from discovery to clinic. J Ethnopharmacol 51(1–3):239–253 (discussion 253–234)
Basili S, Moro S (2009) Novel camptothecin derivatives as topoisomerase I inhibitors. Expert Opin Ther Pat 19(5):555–574. doi:10.1517/13543770902773437
Koster DA, Palle K, Bot ES, Bjornsti MA, Dekker NH (2007) Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature 448(7150):213–217. doi:10.1038/nature05938
Lavergne O, Demarquay D, Kasprzyk PG, Bigg DC (2000) Homocamptothecins: E-ring modified CPT analogues. Ann N Y Acad Sci 922:100–111
Zunino F, Dallavalleb S, Laccabuea D, Berettaa G, Merlinib L, Pratesi G (2002) Current status and perspectives in the development of camptothecins. Curr Pharm Des 8(27):2505–2520
Adams DJ, Wahl ML, Flowers JL, Sen B, Colvin M, Dewhirst MW, Manikumar G, Wani MC (2006) Camptothecin analogs with enhanced activity against human breast cancer cells. II. Impact of the tumor pH gradient. Cancer Chemother Pharmacol 57(2):145–154. doi:10.1007/s00280-005-0008-5
Burris HA, Rothenberg ML, Kuhn JG, Von Hoff DD (1992) Clinical trials with the topoisomerase I inhibitors. Semin Oncol 19(6):663–669
Swami U, Goel S, Mani S (2013) Therapeutic targeting of CPT-11 induced diarrhea: a case for prophylaxis. Curr Drug Targets 14(7):777–797
Dallavalle S, Ferrari A, Biasotti B, Merlini L, Penco S, Gallo G, Marzi M, Tinti MO, Martinelli R, Pisano C, Carminati P, Carenini N, Beretta G, Perego P, De Cesare M, Pratesi G, Zunino F (2001) Novel 7-oxyiminomethyl derivatives of camptothecin with potent in vitro and in vivo antitumor activity. J Med Chem 44(20):3264–3274
Petrangolini G, Pratesi G, De Cesare M, Supino R, Pisano C, Marcellini M, Giordano V, Laccabue D, Lanzi C, Zunino F (2003) Antiangiogenic effects of the novel camptothecin ST1481 (gimatecan) in human tumor xenografts. Mol Cancer Res 1(12):863–870
Bom D, Curran DP, Kruszewski S, Zimmer SG, Thompson Strode J, Kohlhagen G, Du W, Chavan AJ, Fraley KA, Bingcang AL, Latus LJ, Pommier Y, Burke TG (2000) The novel silatecan 7-tert-butyldimethylsilyl-10-hydroxycamptothecin displays high lipophilicity, improved human blood stability, and potent anticancer activity. J Med Chem 43(21):3970–3980
Boven E, Van Hattum AH, Hoogsteen I, Schluper HM, Pinedo HM (2000) New analogues of camptothecins. Activity and resistance. Ann N Y Acad Sci 922:175–177
Van Hattum AH, Schluper HM, Hausheer FH, Pinedo HM, Boven E (2002) Novel camptothecin derivative BNP1350 in experimental human ovarian cancer: determination of efficacy and possible mechanisms of resistance. Int J Cancer 100(1):22–29. doi:10.1002/ijc.10434
Pommier Y, Cushman M (2009) The indenoisoquinoline noncamptothecin topoisomerase I inhibitors: update and perspectives. Mol Cancer Ther 8(5):1008–1014. doi:10.1158/1535-7163.MCT-08-0706
Kummar S, Chen A, Gutierrez M, Pfister TD, Wang L, Redon C, Bonner WM, Yutzy W, Zhang Y, Kinders RJ, Ji J, Allen D, Covey JM, Eiseman JL, Holleran JL, Beumer JH, Rubinstein L, Collins J, Tomaszewski J, Parchment R, Pommier Y, Doroshow JH (2016) Clinical and pharmacologic evaluation of two dosing schedules of indotecan (LMP400), a novel indenoisoquinoline, in patients with advanced solid tumors. Cancer Chemother Pharmacol 78(1):73–81. doi:10.1007/s00280-016-2998-6
Antony S, Agama KK, Miao ZH, Takagi K, Wright MH, Robles AI, Varticovski L, Nagarajan M, Morrell A, Cushman M, Pommier Y (2007) Novel indenoisoquinolines NSC 725776 and NSC 724998 produce persistent topoisomerase I cleavage complexes and overcome multidrug resistance. Cancer Res 67(21):10397–10405. doi:10.1158/0008-5472.CAN-07-0938
Baldwin EL, Osheroff N (2005) Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents 5(4):363–372
Vogelzang NJ, Raghavan D, Kennedy BJ (1982) VP-16-213 (etoposide): the mandrake root from Issyk-Kul. Am J Med 72(1):136–144
Ross W, Rowe T, Glisson B, Yalowich J, Liu L (1984) Role of topoisomerase II in mediating epipodophyllotoxin-induced DNA cleavage. Cancer Res 44(12 Pt 1):5857–5860
Lee K-H, Xiao Z (2011) Podophyllotoxins and analogs. In: Anticancer agents from natural products, Second Edition. CRC Press, pp 95–122. doi:10.1201/b11185-6
Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET (2012) Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 18(11):1639–1642. doi:10.1038/nm.2919
Khiati S, Dalla Rosa I, Sourbier C, Ma X, Rao VA, Neckers LM, Zhang H, Pommier Y (2014) Mitochondrial topoisomerase I (top1mt) is a novel limiting factor of doxorubicin cardiotoxicity. Clin Cancer Res 20(18):4873–4881. doi:10.1158/1078-0432.CCR-13-3373
Douarre C, Sourbier C, Dalla Rosa I, Brata Das B, Redon CE, Zhang H, Neckers L, Pommier Y (2012) Mitochondrial topoisomerase I is critical for mitochondrial integrity and cellular energy metabolism. PLoS ONE 7(7):e41094. doi:10.1371/journal.pone.0041094
Faulds D, Balfour JA, Chrisp P, Langtry HD (1991) Mitoxantrone. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in the chemotherapy of cancer. Drugs 41(3):400–449
Parker BS, Cullinane C, Phillips DR (1999) Formation of DNA adducts by formaldehyde-activated mitoxantrone. Nucleic Acids Res 27(14):2918–2923
Boland MP, Fitzgerald KA, O’Neill LA (2000) Topoisomerase II is required for mitoxantrone to signal nuclear factor kappa B activation in HL60 cells. J Biol Chem 275(33):25231–25238
Larsen AK, Escargueil AE, Skladanowski A (2003) Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol Ther 99(2):167–181
Erlichman C, Boerner SA, Hallgren CG, Spieker R, Wang XY, James CD, Scheffer GL, Maliepaard M, Ross DD, Bible KC, Kaufmann SH (2001) The HER tyrosine kinase inhibitor CI1033 enhances cytotoxicity of 7-ethyl-10-hydroxycamptothecin and topotecan by inhibiting breast cancer resistance protein-mediated drug efflux. Cancer Res 61(2):739–748
Stewart CF, Leggas M, Schuetz JD, Panetta JC, Cheshire PJ, Peterson J, Daw N, Jenkins JJ 3rd, Gilbertson R, Germain GS, Harwood FC, Houghton PJ (2004) Gefitinib enhances the antitumor activity and oral bioavailability of irinotecan in mice. Cancer Res 64(20):7491–7499. doi:10.1158/0008-5472.CAN-04-0096
Pommier Y, Huang SY, Gao R, Das BB, Murai J, Marchand C (2014) Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair (Amst) 19:114–129. doi:10.1016/j.dnarep.2014.03.020
Santi DV, Schneider EL, Ashley GW (2014) Macromolecular prodrug that provides the irinotecan (CPT-11) active-metabolite SN-38 with ultralong half-life, low C(max), and low glucuronide formation. J Med Chem 57(6):2303–2314. doi:10.1021/jm401644v
Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB Jr, Stewart L (2002) The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci USA 99(24):15387–15392. doi:10.1073/pnas.242259599
Acknowledgements
We would like to thank the past and current Bjornsti and van Waardenburg lab members, and colleagues for inspiring discussions that contribute to the continued development of the DNA topoisomerases field. Our apologies to colleagues whose work we did not mention, due to space limitations. RCAMvW greatly appreciates the financial support from the Department of Pharmacology and Toxicology, UAB ACS-IRG Junior Faculty Development Grant (ACS-IRG-60-001-53), the UAB Comprehensive Cancer Center Faculty Development Grant, and DOD OCRP pilot award W81XWH-15-1-0198. M-AB acknowledges the support from the National Institutes of Health and, along with RCAMvW, support from the Alabama Drug Discovery Alliance and the Cancer Center Core Support Grant P30CA013148.
Funding
This study was in part funded by American Cancer Society UAB ACS-IRG Junior Faculty Development Grant (ACS-IRG-60-001-53), Department of Defense OCRP pilot award W81XWH-15-1-0198 to RCAMvW, and the National Institutes of Health Cancer Center Core Support Grant (P30CA013148) to RCAMvW and M-AB.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Cuya, S.M., Bjornsti, MA. & van Waardenburg, R.C. DNA topoisomerase-targeting chemotherapeutics: what’s new?. Cancer Chemother Pharmacol 80, 1–14 (2017). https://doi.org/10.1007/s00280-017-3334-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-017-3334-5