
Abstract
Since the late 1990s, there has been rapid multiplication of data on the anti-cancer properties of artemisinins. This article reviews the status of progress of artemisinin and its derivatives as anti-cancer agents in clinical trials, case reports, and in vitro/in vivo studies. Particular attention is laid on the combinations of artemisinins and synthetic chemodrugs to enhance the latter’s efficacy. An attempt is here made to rationalize the synergistic effects of a few common anti-cancer drugs of the anthracycline, taxane, anti-metabolite, and platinum-based drug families. The various pathways that mediate the action of artemisinins as reported over the past decade are here summarized highlighting also the biomarkers that could be used to better predict the efficacy of the sesquiterpenoids. Their main action seems to be directed toward stalling tumor cell proliferation through cell cycle arrest mediated by reactive oxygen species (ROS). The emergence of artemisinins’ nano-based formulations in combination with chemodrugs to enhance drug bioavailability and targeting as well as immunotherapy is also reviewed. The enhanced efficacy of artemisinin dimers compared to the parent molecules and standard chemotherapy is analyzed. While these therapies hold promises, it may be premature to conclude on their efficacy in the absence of clinical studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Artemisinin (ART) (Fig. 1) extracted from Artemisia annua was in the spotlight in a recent past as Youyou Tu was awarded half the 2015 Nobel Prize in Physiology or Medicine for her discovery of that compound as a novel therapy against malaria, as approved in 1986. Tu and co-workers established that artemisinin and dihydroartemisinin also exhibited immunosuppressive and anti-inflammatory effects [1]. In addition to their potency against malaria, several groups have established that artemisinin and its derivatives—artesunate (ARS) and dihydroartemisinin (DHA) (Fig. 1) are also active against cancer cells [2–6].

Artemisinin (ART), dihydroartemisinin (DHA), and artesunate (ARS)
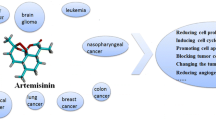
A PubMed search yielded 188 papers on anti‑cancer effects of ART, 75 papers on ARS, 71 papers on DHA, 3 papers on artemether, and 2 on artemisone spanning a period of 20 years. Most in vitro and animal studies have reported on beneficial outcome but some studies have shown no major therapeutic improvement. An exhaustive study performed in 2015 on 55 cancer cell lines reports on the inhibitory effects against various cancers—pancreatic, osteosarcoma, lung, colon, melanoma, breast, ovarian, prostate, central nervous system, lymphoma, leukemia, and renal [7]. The interest for artemisinin and its derivatives resides in their safe toxicity profile.
However, data on human cancer is still lacking. Only a few case reports and clinical trials can be found in literature and the water-soluble ARS was used in most of them. It has shown good tolerability, is easily administered, and lacks serious side effects which can facilitate prospective randomized trials so that artemisinins do not end up in the basket of molecules with a promising panoply of in vitro and animal studies but not enough extensive clinical studies for their clinical application in cancer therapy.
One of the main problems with artemisinin is its low water solubility and poor bioavailability with a short half-life in vivo of ∼2.5 h. DHA, with a half-life ~2 h, is considered as the main active metabolite of artemisinins [8]. To overcome the solubility problem, other semi-synthetic derivatives such as ARS, artemether, and artemisone have been developed.
This review focuses on the outcomes of clinical trials and in vitro/in vivo studies where artemisinins have been used alone or in combination with chemodrugs to fight cancer. In addition, we summarize the different pathways for the anti-cancer action of artemisinins and attempt to rationalize their synergistic effect with common chemodrugs. In a last section, emerging artemisinin-based therapies such as nanotherapy and immunotherapy are discussed.
Clinical studies involving artemisinins
Clinical trials
Krishna et al. [9] reported on the use of oral ARS in a single center, randomized double-blind, placebo-controlled trial targeting colorectal cancer (CRC). Patients (n = 23) received preoperatively either 14 daily doses of oral ARS (200 mg; n = 12) or placebo (n = 11) with medication stopped 48–72 h prior to surgery. Apoptosis in >7% of cells was seen in 67 and 55% of patients in ARS and placebo groups, respectively. Probabilities of ARS reducing Ki67 and increasing CD31 expression were 0.89 and 0.79 using Bayesian analysis. During a median follow-up of 42 months, 1 patient in the ARS and 6 patients in the placebo group developed recurrent CRC. The conclusion of this study was that ARS has anti-proliferative properties in CRC and is generally well tolerated. CD31 shows the vascular status of CRC but do not indicate the angiogenic intensity. A study showed that an important number of vessels (around 40%) in the CRC tumor area are neoformation vessels where CD105 is expressed and which increases with the neoangiogenic progression [10]. In this clinical trial, CD31 is probably not promoting angiogenesis, thus increasing the anti-proliferative effect of ARS.
In an open-label single-center study to assess the efficacy, safety, and clinical benefit of oral artenimol-R (DHA is commonly called artenimol, R = hemi-succinate ester) on advanced cervical carcinoma, remission of patients was shown [11]. Ten patients were enrolled in the study with an initial treatment phase of 28 days and a follow-up phase. Patients were given in the initial treatment phase 100 mg/day (two tablets of 50 mg) of DHA-R during week 1, this was increased to 200 mg/day (1 tablet) for the rest of the 28 days. In the follow-up phase, patients received 200 mg/day for another 28 days. No further treatment was administered and patients were followed-up until their death. Out of the 10 patients, one patient improved sufficiently after the first treatment to allow the removal of the primary tumor and six patients were in remission as at August 2010. It is to be noted that the usual prognosis of patients with metastasized cervical carcinoma, is a survival time of about 4 months. No adverse effect occurred during the treatment and immunohistochemistry showed downregulation of p53, EGFR, Ki-67, and CD31, while the expression of transferrin receptor protein 1 (CD71) increased. The downregulation of p53 and EGFR indicated reversing of the malignant features of tumor, Ki-67 indicated slowing down of tumor proliferation, and CD31 showed reduced angiogenesis. These showed that DHA-R is probably acting via induction of oxidative stress which lead to cell cycle arrest and inhibition of angiogenic proteins.
The anti-cancer combination vinorelbine and cisplatin was used with ARS in the treatment of advanced non-small cell lung cancer in a study consisting of 120 patients [12]. The group was divided randomly into control group [n = 60, vinorelbine (25 mg/m2, once-a-day intravenous injection, at the 1st and 8th day)] and cisplatin (25 mg/m2, once-a day intravenous drip, at the 2nd to 4th day) and ARS with chemotherapy group (trial group, n = 60, chemotherapy regimen was the same as control group). Daily ARS intravenous injections (120 mg, from the 1st day to 8th day, for 8 days) were administered to patients with at least two 21-day cycles of treatment. Disease controlled rate (DCR) and time to progression (TTP) showed significant difference between the trial group (DCR: 88.2%, TPP: 24 weeks) and the control group (DCR: 72.7%, TPP: 20 weeks) (P < 0.05). The short-term survival rates were 45.1 and 34.5% and 1-year survival rates were 45.1 and 32.7% between the trial group and the control group, and, respectively. This study concluded that ARS combined with the chemotherapy regimen can reduce the proliferation of the disease without additional side effects.
Artemisinins’ half-life in vivo is an important parameter for their clinical efficacy. One recent study looked at the population pharmacokinetics of ARS and the active metabolite DHA for daily administration in metastatic breast cancer patients [13]. Twenty-three patients participated in the study receiving 100, 150, or 200 mg of oral ARS and DHA. Saliva drug and plasma concentrations were measured. A direct agreement between estimated DHA saliva/plasma ratio and reported DHA unbound fraction in human plasma was found. ARS saliva concentrations showed a poor correlation with plasma concentrations which may be a result of poor bioavailability.
Some reports have expressed concerns about the neurotoxicity of ART impairing the auditory system [14]. The safety for hearing of ARS was investigated in a phase I open-label, uncontrolled, monocentric, dose-escalation study (over 4 weeks), as an add-on therapy in 23 patients with advanced breast cancer [15]. Patients received either 100, 150, or 200 mg oral ARS daily in addition to their on-going chemotherapy. The main outcome was that ARS did not show any dose-limiting auditory toxicity.
Case reports
Case reports using artemisinins have so far dealt with patients receiving treatment on a compassionate basis upon failure of standard chemotherapy, surgery, and/or radiotherapy.
Artemisinin and derivatives
A patient with prostate carcinoma [pT3bN1M1, Gleason score 8 (4 + 4)], and a prostate specific antigen (PSA) level >800 µg/l was given long-term oral treatment (approximately 7 months) with A. annua capsules (continuously 5 × 50 mg/day) after a prior treatment with bicalitumide [16]. The PSA level dropped to 0.98 µg/l at the end of 5 weeks treatment and then both PSA and ostase levels increased steadily. After an additional 6 months, PSA level continued to increase, indicating tumor recurrence and skeletal metastases. The patient was then placed on ARS injections (2 × 150 mg twice weekly) but PSA and ostase levels rose to 1245 µg/l and 434 U/l, respectively, and MRT revealed the tumor acquired resistance. This study concluded that ART required long-term treatment.
Singh and Verma [17] used ARS injections and tablets to treat a 72-year-old male patient suffering from laryngeal squamous cell carcinoma. The patient received daily injection of 60 mg of ARS from day 1 to day 15. One tablet of 50 mg ARS was taken orally as from day 16 onward. The patient also received a daily oral dose of ferrous sulfate (150 mg) and folic acid (0.5 mg). This treatment was administered over a period of 9 months. The tumor was significantly reduced (by 70%) after 2 months of treatment with improved quality of life (gain in weight and physically stronger). The patient lived for nearly 1 year and 8 months (until his death due to pneumonia).
A case report presented by Singh and Panwar [18] describes the use of artemether on a 75-year-old male patient with pituitary macroadenoma. The treatment for non-secreting macroadenoma are invasive (transsphenoidal surgery) and have significant side effects (radiation). The rationale for using artemether was to have an alternative treatment that has minimal side effects. Artemether was administered orally to the patient over a period of 12 months. No reduction in tumor size was detected but CT scan indicated reduction in its density (72–77 HU in the first scan vs 51–59 HU in the last scan). The authors proposed that the remaining mass may consist mostly of dead cells, which in older patients takes a longer time to be scavenged by microglia and macrophage cells. The clinically related symptoms and signs were significantly resolved improving the patient’s quality of life.
Some case reports have been mentioned by Rowen [19] where patients were either cancer free or have experienced regression at the end of artemisinins’ treatment for the following: non-Hodgkin’s lymphoma/artesunate (60 mg, 14 days); non-resectable non-small cell lung cancer carcinoma/artemisinin (500 mg); metastatic stage 4 breast cancer/artesunate/artemisinin (300 mg); multiple skin cancer/topical artemisinin; metastatic breast cancer/artemisinin (oral); and metastatic ovarian cancer/artemisinin (400 mg).
Artemisinins in combination therapy
The first report on ARS combination with standard chemotherapy on two cancer patients was published in 2005 [20]. These patients suffered from metastatic uveal melanoma where standard chemotherapy alone was ineffective in stopping tumor growth. One patient experienced a temporary response after the addition of ARS to fotemustine. There was regression of some lung metastases but development of new CNS tumors. The patient survived 11 months after the combination treatment which is longer than the median survival time. The second patient first experienced a stabilization of the disease after the addition of ARS to dacarbazine, followed by objective regressions of splenic and lung metastases. This patient was still alive 47 months after first diagnosis of stage IV uveal melanoma (median survival of 2–5 months).
A study from Vietnam, reported that 50–60% of 400 cancer patients have achieved long-term remission using ART with a comprehensive integrative cancer strategy. The author reported on the case of a 47-year-old female with terminal liver cancer who has attained complete remission after two and half years treatment with artemisinin. He compared cancer to the malaria parasite where prolonged treatment prevented relapse [21].
Recently, a 52-year-old male patient suffering from glioblastoma multiforme, where further tumor-specific treatment was unlikely to be successful, was administered a combination of dichloroacetate (DCA) and ARS on a compassionate basis by an alternative practitioner [22]. Prior treatment of the patient was a surgery followed by radiotherapy. The DCA and ARS treatment started 148 days after surgery. However, the patient died 9 days later of fatal liver and bone marrow toxicity. The amount of DCA used was unknown and ARS dose was 2.5 mg/kg bodyweight, both administered intravenously while the patient had a stable/unchanged concomitant medication. DCA is known to be hepatotoxic but the report could not correlate it with patient’s death as the dose applied was not disclosed while hepatotoxicity due to ARS is very rare even in malaria patients where larger clinical trials have been conducted. This case report presents a word of caution on the compassionate use of DCA/ART combination therapy outside of clinical trials.
Artemisinin combinations: exploiting synergistic effect
A number of combinations have been reported between artemisinins and synthetic chemodrugs [8]. These studies have shown both additive synergistic and adverse effect of artemisinins on chemotherapy. Doxorubicin, gemcitabine, paclitaxel, and cisplatin are among the most potent drugs used in chemotherapy for a wide range of cancer. Many strategies are targeted toward enhancing their efficacy and their combination with artemisinins is also directed toward the same goal. Artemisinin is believed to be more efficient in terms of targeting cancer cells due to their high intracellular iron levels.
Artemisinins and Anthracyclines (Doxorubicin, pirarubicin)
Mitochondrial depolarization
Chemosensitization is proposed as one mechanism through which artemisinins exert their synergistic effect. For instance, ART, ARS, or DHA in combination with pirarubicin or doxorubicin increased the efficacy of both pirarubicin and doxorubicin on MDR cell lines [23]. Pirarubicin and doxorubicin are anthracyclines, the efficacy of which depends on their intracellular concentration. The artemisinins did not act by decreasing the Pgp- and multidrug resistance-associated protein 1 (MRP1)-mediated efflux of the drugs but instead decreased the mitochondrial potential and ATP concentration. The latter is required in higher amount in resistant cells. It is interesting to note that in this study ARS and DHA showed 15-fold greater efficacy than ART in K562 cells and 60-fold greater efficacy in GLC4 cells.
ARS was shown to induce apoptotic cell death in doxorubicin-resistant cell line (T-cell leukemias). In combination with doxorubicin, it synergistically enhances doxorubicin-induced apoptosis due to different drug mechanism pathways [24]. ARS acts through the mitochondrial intrinsic pathway through generation of ROS, whereas doxorubicin acts via inhibition of DNA polymerases, DNA topoisomerase II, and DNA methyltransferase. This study suggested that ARS can act synergistically with other DNA intercalating anti-cancer drugs.
The combination of DHA and doxorubicin on breast cancer cells (MCF-7) showed anti-proliferative effect and DOX-mediated apoptosis. Again DHA did not influence accumulation of DOX in cells but decreased mitochondrial potential and activated caspase cascades [25]. Mitochondrial depolarization was only moderate with DHA or DOX alone, however, a combination of both resulted in high depolarization.
SERCA inhibitors reducing toxic effect of doxorubicin
ART is known to act as SERCA (sarcoplasmic/endoplasmic reticulum Ca2+-ATPase) inhibitor in Plasmodium falciparum [26]. In human colon cancer cells (HT29), it reduced the activity of SERCA and increased cytosolic [Ca2+] [27]. These two resulted in the cells becoming more resistant to the toxic effects of doxorubicin when used in combination with ART. The increase in Pgp protein and Ca2+ caused a decrease in intracellular doxorubicin accumulation. The rise in Ca2+ was thought to activate Ca2+/calmodulin-dependent protein kinase II (CaMKII), which in turn activates the transcription factor HIF-1α resulting in overexpression of Pgp.
Binding with Translationally Controlled Tumor Protein (TCTP)
TCTP is one of the targets for both tumor cells and plasmodia. A decrease TCTP protein expression is critical for tumor growth/survival [28]. Apoptosis may result from the reduction of phospho-TCTP levels by DHA without excluding ROS mediated action in triple-negative breast cancer [29]. In combination with doxorubicin, an alternative pathway to inhibit the progression is obtained by regulating the phospho-TCTP function. Lower chemotherapeutic doses of doxorubicin were required and this is expected to reduce side effects in patients.
Artemisinins and Anti-metabolites (Gemcitabine)
Reduction of transcription factor nuclear factor-kB (NF-kB) activation
DHA was shown to promote the anti-cancer effect of gemcitabine in vitro (BxPC-3 and PANC-1 pancreatic cancer cell lines) and in vivo (BxPC-3 tumor-bearing mice) [30]. Transcription factor nuclear factor-kB (NF-kB) is constitutively activated in about 70% of pancreatic adenocarcinomas which inhibits apoptosis and promotes proliferation. Gemcitabine is known to induce NF-kB activation and the presence of DHA prevents this activation, thus tumor growth was inhibited by 45% with the DHA/gemcitabine combination.
ART/gemcitabine only reduced proliferation of hepatoma cells in HepG2 xenograft tumor slightly compared to ART or gemcitabine alone. However, combination of gemcitabine and DHA led to a significant decrease in cell survival [31]. Gemcitabine alone decreased tumor growth by 34.9% while in combination with ART or DHA, the decrease was 62.3 and 78.4%, respectively. The in vivo activities of the artemisinins were associated with their capacity to induce G1-phase arrest and apoptosis which was additive to gemcitabine action.
Role of ROS
A ROS-independent synergistic action between DHA and gemcitabine was shown in a non-small cell lung cancer cell line (A549 cells) [32]. Cell growth was inhibited through apoptosis. The study showed that the combination of gemcitabine and DHA did not increase ROS significantly compared to either drug alone. The proapoptotic Bcl-2 antagonist killer 1 (Bak)-mediated intrinsic apoptosis pathway, loss of mitochondrial potential, and the Fas-caspase-8-mediated extrinsic apoptosis pathway were responsible for the synergistic action.
As a follow-up, the proapoptotic mechanisms of DHA in gemcitabine-resistant A549 (A549GR) cells were studied by Zhao et al. [33]. The cells have the following characteristics: low basal antioxidant capacity and higher level of basal ROS and intracellular Fe2+. These gemcitabine-resistant cells showed similar ROS production with gemcitabine than A549 cells but more rapid ROS generation with DHA. Bcl-2-like protein 4 (Bax) activation, loss of mitochondrial potential, caspase activation, and apoptosis were enhanced. DHA had more potent proapoptotic actions via ROS-dependent apoptotic pathway involving Bax and caspases.
ART and artemisone’s abilities to sensitize MCF7 cells to gemcitabine have been evaluated [34]. In this study, artemisone alone was found to effect cell cycle arrest 30-fold more than ART. Artemisone effected G1-selective blockade, whereas ART resulted in a general arrest in all phases. In combination, ART interfered negatively with the action of gemcitabine while artemisone had no antagonistic effect but did not show any additive effect.
Artemisnins and Taxanes (Paclitaxel)
In a spirit to provide a firmer basis for deciding on synergy between artemisinins and anti-cancer drugs, a cancer line panel (91 cell lines) comprising conventional commercially available cell lines as well as those derived directly from patient tumor xenografts was used. The efficacy of ARS, DHA, and artemisone as anti-cancer drugs and their potential for synergies with established anti-cancer drugs were tested [35]. It was found that paclitaxel clustered with the three artemisinins. The latter stop mitosis at the G1/G2 phase, while paclitaxel does so at the metaphase.
Inhibition of Raf/MEK/MAPK signaling pathway
DHA/paclitaxel combination was used on ovarian cancer cell lines SKOV3 and OVCAR3. DHA synergized with paclitaxel (CI value 0.6–0.73) in SKOV3 [36]. DHA acts by cell arrest in G2/M phase and downregulated oncogenic transcription factor FOXM1 expression. FOX1 expression peaks at the G2/M phase of the cell cycle [37]. For nuclear translocation to occur, phosphorylation of FOXM1 is required and this occurs through activity of the Raf/MEK/MAPK signaling pathway. DHA inhibits serum-induced phosphorylation of MAPK, and thus Raf/MEK/MAPK signaling pathway. DHA targeting of FOXM1 induces apoptosis and stabilizes microtubule dynamics, thus sensitizing tumor cells to paclitaxel-induced apoptosis. Paclitaxel acts by promoting the assembly of microtubules and blocking cells in the G2/M phase which then become unable to form a normal mitotic apparatus [38].
Artemisinins and Platinum-based drugs (cisplatin)
Synergy through ROS
Using MGC-803 gastric cancer cell line, ART combined with cisplatin was investigated for synergistic epithelial–mesenchymal transition, angiogenesis, and ATP generation [39]. ATP generation as well as VEGFA, VEGFB, VEGFC, N-cadherin, and Vimentin levels were lower than those of the individual drugs. Early apoptosis rate, late apoptosis rate, and E-cadherin level were also higher than the individual drugs. The synergy was attributed to the generation of ROS.
Downregulation of RAD51
ARS acts on cancer cells by oxidative stress and DNA double-strand breaks (DSBs). It has also been shown to downregulate RAD51, essential for DNA homologous recombination, which impairs DSB repair in ovarian cancer cells [40]. Most patients with advanced ovarian cancer undergo surgical resection followed by platinum-based chemotherapy. Relapse with platinum-resistant disease is common. ARS has been shown to sensitize ovarian cancer cells resistant to cisplatin through the downregulation of RAD51. ARS and cisplatin can both induce DSBs, and together they acted synergistically. They prevented the clonogenic formation of ovarian cancer cells while on their own they showed mild inhibitory effect on the colony-forming ability of ovarian cancer cells.
Deactivation of mTOR kinase
Cisplatin is the most efficient drug in the treatment of ovarian cancer but many tumors become refractory to cisplatin treatment over time. Abnormal activation of mTOR phosphorylation was found in cisplatin-resistant ovarian cancer cells (SKOV3/DDP) after cisplatin monotherapy [41]. Cisplatin/DHA combination inhibited proliferation in SKOV3/DDP cells. This mechanism was attributed to DHA deactivation of mTOR kinase and promotion of apoptosis. It was also found that inhibition of autophagy did not affect SKOV3/DDP cells’ survival in the presence of cisplatin. Apoptosis and autophagy can be synergistic or antagonistic depending on the cancer cell type and/or stimuli, and this needs further investigation.
Inhibition of angiogenesis
DHA on its own can inhibit vascular endothelial growth factor (VEGF) expression in tumor cells [42]. DHA-cisplatin combination yielded regression of tumor size in Lewis lung carcinoma xenografted C57BL/6 mice [43]. Metastasis was also completely inhibited by the combination. Inhibition of angiogenesis via downregulation of VEGF receptor resulting in apoptosis was proposed to be the combined mechanism of action.
Based on the experimental data here discussed, we have attempted to match the cell cycle targets of artemisinins with classical chemo drugs as depicted in Scheme 1 for successful synergistic combination. Artemisinins have very low toxic effects for patients and their combination with classical chemotherapy can reduce the harmful side effects in normal tissues. However, these data are yet to be confirmed in clinical trials.

Matching the cell cycle targets of artemisinins with anti-metabolites (gemcitabine), anthracyclines (doxorubicin), taxanes (paclitaxel), and platinum-based (cisplatin) chemodrugs
Artemisinins’ mechanism of action
Over the past 5 years, various reviews have reported on artemisinins’ anti-cancer mechanism of action [7, 8, 44, 45]. We summarize here the main pathways that mediate that action based on in vitro and in vivo data. It is now accepted that cleavage of the endoperoxide bridge of artemisinins is the major trigger for their therapeutic action which is thought to proceed via (1) apoptosis, ferroptosis [46], or necrosis; (2) anti-angiogenesis [47, 48]; (3) oxidative stress; (4) tumor suppressor genes; and (5) protein targeting [49]. ART has been shown to act on the G1 phase [50–52], and DHA and ARS on the G2/M phase arrest [53, 54]. G0/G1 phase arrest has also been reported for ARS in the presence of iron [55], G1 arrest [56, 57], and G0/G1 [58] have been reported for DHA. These data indicated that cell cycle arrest may be cell type dependent, dose dependent or dependent on the type of chemotherapeutic combination used, as discussed in the previous section. Under hypoxic conditions, low-dose DHA caused higher inhibition, whereas high doses (required for anti-angiogenic or anti-tumor activity) were more effective in normoxia [59]. Table 1 lists some of the pathways shown on various cancer cell lines and their relationship with cell cycle arrest.
Based on experimental data, Scheme 2 summarizes the possible pathways that regulate the action of artemisinins (ARTs). Cancer cells are more prone to cytotoxic effect of artemisinins due to their high intracellular iron levels, which is essential for rapid cell division and proliferation. Once artemisinins are inside the cells, iron will cleave the endoperoxide bond resulting in the formation of oxygen and alkylating carbon radicals. Tumor cells have a high O2 −:H2O2 ratio and these give rise to hydroxyl radical (·OH) which is responsible for most of the oxidative damages attributed to ROS. The artemisinin radicals activate lysosomes [75] and affect the electron transfer chain in the mitochondria [76] causing a breakdown in transmembrane potential, release of cytochrome c, caspase activation, and DNA damage. Lysosomal iron thus mediates artemisinin-induced mitochondrial ROS production. The generated oxidative stress plays a central role in cell cycle arrest, reduced proliferation, anti-angiogenesis, apoptosis, autophagy, ferroptosis, and inhibition of angiogenesis. The predominance of one pathway or another will depend on cancer type, expression of antiapoptotic or proapoptotic genes, signaling pathways, or downregulation of growth factors. In general, low dosage of artemisinins seems to favor cell cycle arrest, whereas higher dosage may result in apoptosis [8, 71, 77].

Overview of the major pathways for artemisinins’ action on tumor cells resulting in anti-angiogenesis, apoptosis, autophagy, cell cycle arrest, ferroptosis, and reduced proliferation
ROS regulate mitogen-activated protein kinase (MAPK)/Erk cascade, phosphoinositide-3-kinase (PI3K)/Akt-regulated signaling cascades, and the IκB kinase (IKK)/nuclear factor κ-B (NF-κB)-activating pathways to promote cell proliferation, nutrient uptake, and cell survival in tumors [78]. In cancer cells, a redox balance is maintained between high rate of ROS production and high rate of antioxidant activity as uncontrolled ROS levels lead to oxidative stress-induced cell death. Low expression of antioxidant enzymes in cancer cells make them again more selective for artemisinins’ cytotoxic ROS action. The site of ROS production within cells determines whether damage or redox signaling occurs [79]. Oxidative stress leads to damaged mitochondria which is eliminated by autophagy, thus decreasing proliferation [80]. ROS increase does enhance autophagy but at the same time it promotes cancer cell drug resistance and cancer cell survival. The relationship between ROS and autophagy appears key in understanding the action of artemisinins, in particular their short- v/s long-term use, their dosage or their synergistic effect in combination with other drugs.
Artemisinins can activate or inhibit multiple cell signaling pathways [81]. Microarray data showed that five key signaling pathways: PI3K-Akt, T-cell receptor, Toll-like receptor, TGF-β, and insulin signaling pathways were involved in artemisinin-mediated anti-cancer effects [82]. TGF-β and PI3K-Akt act on cell cycle and apoptosis, and immunity is possibly regulated by T-cell receptor and Toll-like receptor. For instance, DHA-induced apoptosis in prostate cancer cells is attributed to DHA action on PI3K-Akt pathway [83]. Another possible contribution of artemisinins lies in the inhibition of NO production. The latter regulates tumor growth, migration, invasion, survival, angiogenesis, and metastasis in a concentration-dependent manner. ARS showing the highest ability of NO inhibition probably related to its solubility [84].
New directions
Artemisinin and its derivatives do not exert any major toxic side effects. Prolonged use appears to yield better results as reported in the few available clinical trials. In vitro studies show that they enhance the efficacy of the most commonly used chemo drugs. However, artemisinin has a short in vivo half-life, a problem for clinical application. Pioneering work in enhancing the therapeutic effect of artemisinins includes artemisinin tagged to iron-delivery proteins and synthesis of artemisinin dimers [85]. Artemisinin-transferrin conjugate has been shown to be more potent than artemisinin in killing cancer cells [86]. However, the artemisinin–transferin complexes might not exhibit the anti-angiogenesis, anti-inflammatory, and anti-metastasis properties of artemisinin as these are not mediated by transferrin receptor mechanisms.
Recent findings on enhancing the therapeutic effect of artemisinin derivatives such as dimers are discussed. The synthesis of artemisinin dimers enhances the stability and efficacy of the parent artemisinin molecules without enhanced toxicity toward non-diseased cells.
Furthermore, other strategies including nanoformulations and immunotherapy are here reviewed. Nanoformulations could help solve the bioavailability problems to further improve chemotherapeutic outcome. Exploring the capacity of artemisinins’ immunotherapeutic effect can provide more avenues to exploit their therapeutic capacities.
ART-based nano drug delivery systems
Poor delivery and stability of artemisinins reduce their cytotoxic effect. Both can be improved by encapsulating artemisinins into nano drug delivery systems which additionally provide targeting capacity. Only a few in vitro and in vivo studies have been reported on ART-based nano drug delivery systems. These studies have reported mainly on the use of PEG-based and liposomal systems with size in the range of 5 nm–1 µm.
Artemether’s solubility has been increased by 3- to 15-fold using pegylated lysine-based copolymeric dendritic micelles (5–25 nm, loading 0.5-1 g/g) with prolonged release of up to 1–2 days in vitro [87]. ART crystals have been encapsulated with chitosan, gelatin, and alginate (766 nm) with a 90% encapsulation efficiency and improved hydrophilicity [88]. Enhanced dissolution and stability of ART have also been achieved by nano-confinement in ordered mesoporous SBA-15 particles (0.5–1 µm, loading 48.8 ± 1.1 wt%) [89].
A polyethylene glycol–DHA (PEG-DHA) conjugate with moderate drug loadings (2.82, 8.14 wt%) but improved water solubility (82-, 163-fold of DHA) has been reported [90]. The blood circulation half-time was significantly improved (5.75-, 16.75-fold of DHA) and subsequently tumor xenograft assays showed a superior therapeutic effect on inhibition of tumor growth in non-small cell lung cancer compared with native DHA.
DHA-loaded methoxy poly(ethylene glycol)/poly(L-lactic acid) amphiphilic block copolymeric micelles (130 nm) again showed an improved solubility of DHA with a pH-dependent sustained release and higher toxicity on human oral carcinoma KB cells compared to DHA suspension (IC50 = 18.70 and 24.55 µM, respectively) [91].
DHA loaded into conventional (P90G and cholesterol) and stealth liposomes (P90G; cholesterol and PE 18:0/18:0 PEG 2000) (71 and 69% loading) for parental administration showed higher internalization in MCF-7 cells with the conventional liposomes [92].
Phosphatidylcholine, cholesterol, and artemisinin mixed together followed by pegylation yielded pegylated liposomal artemisinin (455 nm, encapsulation efficiency ∼91%) which was tested on MCF-7 breast cancer cells [93]. A decrease in IC50 value from 2.7 ± 0.35 for ART to 1.58 ± 0.3 µg/ml for PEG-Nanoliposomal artemisinin was observed. This result was attributed to the fact that PEG enhanced the drug solubility, as well as its contact with the target cells.
DHA/gelatin and DHA/hyaluronan (30–40 nm) with entrapment efficiencies 13 and 35%, respectively, exhibited greater inhibitory effects on human lung cancer A549 cells compared to DHA alone, this was attributed to the greater aqueous dispersion of DHA [94].
ART loaded into tumor lymphatics-homing peptide (LyP-1) conjugated PEG–PCL micelles (LyP-1-PM) showed higher cellular uptake and higher inhibition effect than PM-artemisinin against MDA-MB-435S breast tumor-bearing mice with delivery to both the tumor and its lymphatics [95].
Nano-vesicular niosomes and solid lipid nanoparticles encapsulated artemisone (67 ± 6%, 211 nm and 79 ± 5%, 295 nm, respectively) showed high toxicity against human melanoma A-375 cells and negligible toxicity toward the normal skin cells (human keratinocytes HaCaT) [96]. Artemisone-loaded nano-vesicles almost completely inhibited the melanoma cells compared to free drug.
Recently, transferrin-eight-arm-polyethylene glycol–dihydroartemisinin nanoparticles (TF-8arm-PEG–DHA NPs) (147 nm, 10% wt drug) have been prepared and tested on Lewis lung carcinoma (LLC) cells and LLC-tumor-bearing mice, which overexpress transferrin receptors (TFRs) [97]. This system was compared to free DHA and non-modified 8arm-PEG–DHA NPs. In vitro studies indicated that in the presence of esterase (abundant in cytoplasm), the nanoparticles were quickly hydrolyzed and released the DHA. The in vitro cytotoxic assay showed more potent effect of TF-8arm-PEG–DHA NPs and their highly selective nature for tumor cells that overexpress TFRs. In vivo studies showed synergistic effect of PEG and TF, resulting in better anti-tumor efficacy (reduced tumor growth) and less side effects compared with free DHA and non-modified NPs.
More such in vivo studies are required to provide measurable data of the benefits of artemisinins’ nano drug delivery systems.
Very recently, a pH sensitive system has been engineered where ART and an iron source were incorporated. ART, as a prodrug, was loaded into the inner space of hollow mesoporous silica (HMS) nanoparticles (NPs) (ART loading 9.6%) [98]. The pore outlets of HMS were then capped with Fe3O4 NPs (ART@HMS-Fe3O4) which were stable with almost no leakage of ART. In the lysosomal compartment (pH 3.8–5.0) of cells, there was sustained release of both ART and Fe3O4 NPs. The latter were metabolized to free iron which resulted in high amounts of free radicals from the released ART. In vitro cytotoxicity assay against ZR75-30 human breast cancer cells revealed promising anti-cancer efficacy of this ART/Fe3O4 co-delivery nanosystem but the safety profile of Fe3O4 nanoparticles has yet to be confirmed in vivo.
Nano drug delivery systems: combination therapy
Combining artemisinins with chemotherapy in nano drug delivery systems can improve efficacy through higher combination index by protecting both drugs from biological factors such as enzymatic degradation, and thus increase the intranuclear accumulation. However, drug PK profiles and the absence of anti-cancer drug-ART interactions will have to be fully established for efficient and safe combination therapy. DHA and hydrophilic doxorubicin co-loaded core–shell-type lipid/particle assemblies (poly(lactic-co-glycolic acid) nanoparticle core coated with a 1,2-dipalmitoyl-sn-glycero-3-phosphocholine shell) (150 nm) resulted in enhanced accumulation of doxorubicin in nuclei of HeLa and HepG2 cancer cells, and thus better treatment efficiency [99]. An interesting aspect of this system is that DHA had a fast release and DOX a slow release providing a time-staggered combination treatment effect. DHA alone has a very low cytotoxicity to both cancer cells (IC50 = ∼20 µg/mL) and DHA/DOX (10:1 w/w) IC50 values were 17- and 9-fold less. This indicated that DHA and DOX had a synergistic effect due to their different targets and faster release of DHA improved DOX cytotoxicity.
Combination of paclitaxel and artemether acting as regulator of apoptosis and inhibitor of brain cancer vasculogenic mimicry (VM) channels have been loaded into multifunctional liposomes [100]. The liposomes could cross the blood–brain barrier through receptor-mediated endocytosis via glucose transporters and adsorptive-mediated endocytosis via electric charge-based interactions. The liposome-loaded artemether/paclitaxel could destroy VM channels, induce apoptosis by activating apoptotic enzymes and proapoptotic proteins and inhibiting antiapoptotic proteins in invasive brain glioma cells and brain glioma-bearing rats.
More potent derivatives: artemisinin dimers
One strategy to enhance the targeting capability and drug efficacy of artemisinin endoperoxides is the synthesis of artemisinin-based dimers [101]. Experimental data reported so far on the use of dimers show effectiveness either by cancer growth inhibition or apoptosis [102, 103]. In transgenic adenocarcinoma of mouse prostate model, some trioxane dimers had high therapeutic index (>150) [104].
The synthetic strategies employed for dimers are symmetric linker dimers, non-symmetric linker dimers, direct reaction of two artemisinin derivatives, and dimers with similar or different artemisinin molecules (Fig. 2). In some cases, the dimers have shown higher anti-cancer efficacy than conventional chemotherapy and the parent molecule. For instance, the symmetric linker-based artemisnin-quinoline dimers exhibited in vitro anti-cancer activity against renal (TK-10), melanoma (UACC62), and breast (MCF-7) cancer cell lines and had 74 times more growth inhibitory effect than the anti-cancer drug etoposide [105]. Artemisinin alcohol and hydrazine dimers were more potent than DHA on rat mammary adenocarcinoma cells (breast tumor MTLn3) in vitro and in vivo where both suppression of cell growth as well as cytotoxicity were observed [106]. A novel artemisinin-derived trioxane diphenylphosphate dimer 838 (ART-838) was shown to reduce cell proliferation and clonogenicity, induced apoptosis, and increased intracellular levels of ROS in 23 tested acute leukemia cell lines [107]. In addition, it was 88-fold more potent than ARS and synergized with established antileukemic drugs and kinase inhibitors to inhibit leukemia cell growth. In immunodeficient NOD-SCID-IL2Rgnull (NSG) mice, ART-838 showed longer plasma half-life than ARS.

Artemisinin-based dimers (X: linker)
The stereochemistry of the dimers appears to play an important role in determining their activity. An α,β ether dimer of DHA was shown to be 20 times more cytotoxic than the β,β ether dimer against EN2 tumor cells [108]. Alcohol, diol, and carboxylic acid trioxane artemisinin dimers were strongly growth inhibitory but not cytotoxic toward several human cancer cell lines [109]. All these dimers have more stable endoperoxide bonds and in the case of carboxylic acid, water solubility. In addition, the water-soluble carboxylic acid derivative is more efficacious than clinically used sodium ARS.
Very importantly, artemisinin dimers do not exert any toxic effect on non-diseased cells like their parent molecules [107] and some of them can be synthesized through a rather simple four-step reaction [109].
Artemisinins: immunotherapy
Chemotherapy can induce immunogenic cell death in addition to the intended therapeutic effect [110]. Harnessing the synergy between immunotherapy and chemotherapy toward additive clinical activity can offer new direction to the use of artemisinins.
Although the action of artemisinins on the immune system is still at embryonic stage, they can effectively exert immunomodulatory effects through three major immune cells namely neutrophils, macrophages, and T cells [111]. Neutrophils and macrophages are inflammatory cells and the latter largely influence the tumor microenvironment (TME) fostering proliferation, survival, and migration [112]. The pro-inflammatory cytokine TNF-α is a key downstream mediator in inflammation. It stimulates proliferation, survival, migration, and angiogenesis in most cancer cells [113]. Tumors can recruit neutrophils and it has been shown in the case of melanoma cells for example that neutrophil-derived TNF stimulates the migration, suggesting that TNF is one factor that neutrophils produce in vivo to initiate metastasis [114]. Extracts from A. annua tested on rat blood inhibited TNF-α production by activated neutrophils in a dose-dependent manner thus exerting anti-inflammatory effect [115].
Tumor-associated macrophages (TAM) inadvertently promote tumor growth and metastasis. In breast cancer, TAMs in the TME play an important role in directing environmental cues for angiogenesis, cell migration, and invasion in preparation for metastasis [116]. TAMs facilitate tumor immune evasion through the secretion of soluble anti-inflammatory cytokines and TNF-α is among the pro-angiogenic growth factors secreted by TAMs. Artemisinins inhibit the secretion of macrophage-derived pro-inflammatory cytokines in particular TNF-α and IL-6 both in vitro (macrophage cells) and in mice [117–119]. In an ovarian cancer model, DHA reduced macrophage infiltration which contributed in preventing metastasis [120].
T-cell recruitment by tumors protects metastatic cancer cells from the immune system [121]. A preclinical study has shown that inhibition of regulatory T cells (Treg) induced immune-mediated rejection of various types of tumors and contributed to tumor regression [122]. ART and DHA suppressed T-cell proliferation and T-cell-related immune response by governing release of the Interleukin-2 (IL-2) in mice model [123]. Artemether also arrested T cells in the G0/G1 phase with reduced IL-2 both in vitro and in vivo using ovalbumin-immunized mice [124]. The immunosuppressive mechanism was shown to proceed via inhibition of Ras-Raf1-ERK1/2 protein kinase cascade activation in T cells. In a T-cell-deficient mice model, artesunate promoted immune restoration [125]. These studies indicate that artemisinins have to be further explored in the treatment of T-cell-mediated immune diseases.
Regulatory T-cells (Treg) contribute to tumor progression through their suppression mechanisms of anti-tumor response of current therapies [126]. Targeting Treg is not an easy task due to the occurrence of adverse reactions, such as hypersensitivity and autoimmunity. In a murine model of breast cancer, ART was found to decrease Treg counts [127]. Delayed-type hypersensitivity was analyzed and the mice treated with 2.16 mg/kg dose of ART responded significantly to antigen challenge.
ARS also exerted the same effect in a mouse model of cervical cancer [128] and in ret-transgenic mouse model of melanoma [129]. Artemether decreased Treg in Balb/c mice bearing 4T1 breast cancer cells [130], whereas DHA showed decrease in Balb/c mice [131].
In the case of cancer, immune therapies try to direct the immune mechanisms to eliminate tumor cells by mainly breaking tolerance and elicit autoimmunity either through antigen-non-specific or antigen-specific approaches [132]. However, this approach results into a number of problems. For instance, administration of interleukin-2 (IL-2) to increase anti-tumor mechanisms resulted in high toxicity such as the vascular leak syndrome. Immunotherapy should not harm the normal immune system. More specific molecules targeting cancer cells and immune cells are required to minimize the unintended side effects of chemotherapy and immunotherapy. In this context, due to their low toxicity toward non-diseased cells, artemisinins have to be further explored. To the best of our knowledge, no clinical study has yet been undertaken on the immunomodulatory effects of artemisinins.
It would also be interesting to distinguish between the chemotherapeutic effect proceeding through the oxidative stress mechanism and the immunomodulatory effect of artemisinins in human studies. The anti-cancer effect of artemisinins proceeds mainly through the generation of ROS via cleavage of the endoperoxide bond. The generation of ROS is the trigger for oxidative stress as described in “Clinical studies involving artemisinins” and “Artemisinin combinations: exploiting synergistic effect” but ROS also have functions in both innate and adaptive immune cells [133]. Elevated ROS in the tumor microenvironment exerts tumor-induced immunosuppressive effect. ROS participate extensively in T-cell activation, apoptosis, and hyporesponsiveness [134]. For instance, they inhibit the proliferation and production of the cytokine IL-2 in T-cells. The major initial source of ROS required for T-cell activation is the mitochondria. The possibility of distinguishing between oxidative stress and immunomodulatory effect of artemisinins can be provided through the careful choice of biomarkers. Oxidative stress status can be evaluated by the assessment of SH-protein, glutathione-S-transferase, and glutathione reductase [135, 136]. Cytokines (IL, TNFα, TGFβ) produced mainly by macrophages and T-cells could be used as biomarkers to assess inflammation [137]. These data would provide valuable information on the mechanism of action of artemisinins to further facilitate transition to clinical trials.
Conclusion
While ruling out toxicity of artemisinins, clinical trials and case reports reported so far seem to show that long-term administration of artemisinins is required for the prevention of recurrence and in vivo residence time is an important parameter for efficacy. Cell cycle arrest, autophagy, and apoptosis are the major pathways mediating the actions of artemisinins. Low dosage of artemisinins seems to favor cell cycle arrest, whereas higher dosage may result in apoptosis. Thus, the dosage and schedule of administration will depend on the tumor stage, status, and progression. The reported dosage in clinical studies and case reports is in the range of 50–200 mg. Case reports where artemisinins are used as sole treatment, seem to favor long-term usage (up to 48 months) which is possible due to negligible toxicity. Shorter treatment schedule (up to 42 days) has been administered when artemisinins are coupled with chemotherapy.
Generation of ROS is the central event cascading into cytotoxic effect of artemisinins. To counter ROS, cells usually make use of antioxidant enzymes. Quantifying these enzymes may act as biomarker to better predict the chemotherapy outcome of artemisinin. The identification of other such measurable biomarkers as p53, P-glycoprotein, transferrin receptor (CD71), vascular endothelial growth factor (VEGF), and von Willebrand factor (CD31) may accelerate clinical application. The ability to use autophagy and ROS as biomarkers in the case of artemisinins will allow better understanding of treatment outcome as ROS accumulation also increases cytoprotective autophagy levels leading to cancer cell drug resistance and cancer cell survival. In general, in vitro studies making use of ART or derivatives singly or in combination have been conclusive. However, the results have yet to be confirmed in vivo. Nanoformulations and artemisinin dimers offer benefits such as enhanced solubility and stability which can improve chemotherapeutic outcome. Better targeting of diseased cells or crossing of blood–brain barrier can also be achieved using nanoformulations. Enhanced solubility of artemisinins via nano drug delivery systems would help in increasing bioavailability, cellular uptake, and in reducing dosage as well as improving toxicity profile and patient compliance. However, in the absence of studies on human subjects, it may be premature to anticipate on outcome and more in-depth studies are needed. More data are also required on the immunomodulatory mechanisms of artemisinins in cancer. Studies are at an embryonic stage but the possibility of coupling the immune and chemo response of artemisinins could be worthy of investigation.
References
Tu Y (2011) The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat Med 17(10):1217–1220
Lai H, Singh NP (1995) Selective cancer cell cytotoxicity from exposure to dihydroartemisinin and holotransferrin. Cancer Lett 91:41–46
Moore JC, Lai H, Li J, McDougall JA, Singh NP, Chou CK (1995) Oral administration of dihydroartemisinin and ferrous sulfate retarded implanted fibrosarcoma growth in the rat. Cancer Lett 98:83–87
Singh NP, Lai H (2001) Selective toxicity of dihydroartemisinin and holotransferrin toward human breast cancer cells. Life Sci 70:49–56
Efferth T, Dunstan H, Sauerbrey A, Miyachi H, Chitambar CR (2001) The anti-malarial artesunate is also active against cancer. Int J Oncol 18:767–773
Singh NP, Lai H (2004) Artemisinin Induces Apoptosis in Human Cancer Cells. Anticancer Res 24:2277–2280
Das AK (2015) Anticancer effect of antimalarial artemisinin compounds. Ann Med Health Sci Res 5(2):93–102. doi:10.4103/2141-9248.153609
Crespo-Ortiz MP, Wei MQ (2012) Antitumor activity of artemisinin and its derivatives: from a well-known antimalarial agent to a potential anticancer drug. J Biomed Biotechnol. doi:10.1155/2012/247597
Krishna S, Ganapathi S, Ster IC, Saeed MEM, Cowand M, Finlayson C et al (2015) A randomised, double blind, placebo-controlled pilot study of oral artesunate therapy for colorectal cancer. EBioMedicine 2:82–90
Deliu IC, Ciurea P, Neagoe D, Bezna MC, Gheonea IA et al (2015) Evaluation of angiogenesis in colorectal cancer. Curr Health Sci J 41(2):145–151
Jansen FH, Adoubi I, J C KC, DE Cnodder T, Jansen N, Tschulakow A, Efferth T (2011) First study of oral Artenimol-R in advanced cervical cancer: clinical benefit, tolerability and tumor markers. Anticancer Res 31(12):4417–4422
Zhang ZY, Yu SQ, Miao LY, Huang XY, Zhang XP, Zhu YP et al (2008) Artesunate combined with vinorelbine plus cisplatin in treatment of advanced non-small cell lung cancer: A randomized controlled trial. J Chin Integr Med 6(2):134–138
Ericsson T, Blank A, von Hagens C, Ashton M, Äbelö A (2014) Population pharmacokinetics of artesunate and dihydroartemisinin during long-term oral administration of artesunate to patients with metastatic breast cancer. Eur J Clin Pharmacol 70(12):1453–1463. doi:10.1007/s00228-014-1754-2
Genovese RF, Newman DB, Brewer TG (2000) Behavioral and neural toxicity of the artemisinin antimalarial, arteether, but not artesunate and artelinate, in rats. Pharmacol Biochem Behav 67(1):37–44
König M, von Hagens C, Hoth S, Baumann I, Walter-Sack I, Edler L et al (2016) Investigation of ototoxicity of artesunate as add-on therapy in patients with metastatic or locally advanced breast cancer: new audiological results from a prospective, open, uncontrolled, monocentric phase I study. Cancer Chemother Pharmacol 77(2):413–427. doi:10.1007/s00280-016-2960-7
Michaelsen F-WS, Saeed MM, Schwarzkopf J, Efferth T (2015) Activity of Artemisia annua and artemisinin derivatives in prostate carcinoma. Phytomedicine 22:1223–1231
Singh NP, Verma KB (2002) Case report of a laryngeal squamous cell carcinoma treated with artesunate. Arch. Oncol 10(4):279–280
Singh NP, Panwar VK (2006) Case Report of a Pituitary Macroadenoma Treated With Artemether. Integr Cancer Ther 5(4):391–394
Rowen RJ (2002) Artemisinin: from Malaria to cancer treatment. Townsend Letter for Doctors & Patients pp 86–88
Berger TG, Dieckmann D, Efferth T, Schultz ES, Funk JO, Baur A et al (2005) Artesunate in the treatment of metastatic uveal melanoma–first experiences. Oncol Rep 14(6):1599–1603
The Cancer Cure Foundation. http://www.cancure.org/12-links-page/43-artemesia. Accessed 11 Oct 2016
Uhl M, Schwab S, Efferth T (2016) Fatal liver and bone marrow toxicity by combination treatment of dichloroacetate and artesunate in a glioblastoma multiforme patient: case report and review of the literature. Front Oncol 6:204–209. doi:10.3389/fonc.2016.00204
Reungpatthanaphong P, Mankhetkorn S (2002) Modulation of multidrug resistance by artemisinin, artesunate and dihydroartemisinin in K562/adr and GLC4/adr resistant cell lines. Biol Pharm Bull 25(12):1555–1561
Efferth T, Giaisi M, Merling A, Krammer PH, Li-Weber M et al (2007) Artesunate induces ROS-mediated apoptosis in doxorubicin-resistant T leukemia cells. PLoS One 2(8):e693. doi:10.1371/journal.pone.0000693
Wu G-S, Lu J-J, Guo J-J, Huang M-Q, Gan L, Chen X-P et al (2013) Synergistic anti-cancer activity of the combination of dihydroartemisinin and doxorubicin in breast cancer cells. Pharmacol Rep 65:453–459
Eckstein-Ludwig U, Webb RJ, van Goethem IDA, East JM, Lee AG, Kimura M et al (2003) Artemisinins target the SERCA of Plasmodium falciparum. Nature 424:957–961
Riganti C, Doublier S, Viarisio D, Miraglia E, Pescarmona G, Ghigo D et al (2009) Artemisinin induces doxorubicin resistance in human colon cancer cells via calcium-dependent activation of HIF-1a and P-glycoprotein overexpression. Br J Pharmacol 156:1054–1066
Lucibello M, Gambacurta A, Zonfrillo M, Pierimarchi P, Serafino A, Rasi G et al (2011) TCTP is a critical survival factor that protects cancer cells from oxidative stress-induced cell-death. Exp Cell Res 317:2479–2489
Lucibello M, Adanti S, Antelmi E, Dezi D, Ciafrè S, Carcangiu ML et al (2015) Phospho-TCTP as a therapeutic target of dihydroartemisinin for aggressive breast cancer cells. Oncotarget 6(7):5275–5291
Wang SJ, Gao Y, Chen H, Kong R, Jiang HC, Pan SH et al (2010) Dihydroartemisinin inactivates NF-κB and potentiates the anti-tumor effect of gemcitabine on pancreatic cancer both In vitro and In vivo. Cancer Lett 293(1):99–108
Hou J, Wang D, Zhang R, Wang H (2008) Experimental therapy of hepatoma with artemisinin and its derivatives: In vitro and in vivo activity, chemosensitization, and mechanisms of action. Clin Cancer Res 14:5519–5530
Zhao C, Gao W, Chen T (2014) Synergistic induction of apoptosis in A549 cells by dihydroartemisinin and gemcitabine. Apoptosis 19(4):668–681
Zhao C, Qin G, Gao W, Chen J, Liu H, Xi G et al (2014) Potent proapoptotic actions of dihydroartemisinin in gemcitabine-resistant A549 cells. Cell Signal 26(10):2223–2233. doi:10.1016/j.cellsig.2014.07.001
Gravett AM, Liu WM, Krishna S, Chan W-C, Haynes RK, Wilson NL et al (2010) In vitro study of the anti-cancer effects of artemisone alone or in combination with other chemotherapeutic agents. Cancer Chemother Pharmacol 67(3):569–577
van Huijsduijnen RH, Guy RK, Chibale K, Haynes RK, Peitz I, Kelter G et al (2013) Anticancer Properties of Distinct Antimalarial Drug Classes. PLoS One 8(12):e82962. doi:10.1371/journal.pone.0082962
Tan X, Chen YI, Chin B, Bieber M, Teng N et al (2014) Artemisinin derivatives synergize with paclitaxel by targeting foxm1 through raf/mek/mapk signaling pathway in ovarian cancer. Abstract 0258, 15th Biennial Meeting of the International Gynecologic Cancer Society, 8–11 November 2014, Australia
Ma RY, Tong TH, Cheung AM, Tsang AC, Leung WY et al (2005) Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J Cell Sci 118(Pt 4):795–806
Weaver BA (2014) How Taxol/paclitaxel kills cancer cells. Mol Biol Cell 25(18):2677–2681. doi:10.1091/mbc.E14-04-0916
Wu M-X (2016) Effect of artemisinin combined with cisplatin intervention on epithelial-mesenchymal transition, angiogenesis and ATP generation in MGC-803 gastric cancer cell lines. J Hainan Med Univer 22(18) (Abstract only available, article in Chinese)
Wang B, Hou D, Liu Q, Wu T, Guo H, Zhang X et al (2015) Artesunate sensitizes ovarian cancer cells to cisplatin by downregulating RAD51. Cancer Biol Ther 16(10):1548–1556. doi:10.1080/15384047.2015.1071738
Feng X, Li L, Jiang H, Jiang K, Jin Y et al (2014) Dihydroartemisinin potentiates the anticancer effect of cisplatin via mTOR inhibition in cisplatin-resistant ovarian cancer cells: Involvement of apoptosis and autophagy. Biochem Biophys Res Commun 444(3):376–381. doi:10.1016/j.bbrc.2014.01.053
Chen H-H, Zhou H-J, Wang W-Q, Wu G-D (2004) Antimalarial dihydroartemisinin also inhibits angiogenesis. Cancer Chemother Pharmacol 53:423–432
Zhou HJ, Zhang JL, Li A, Wang Z, Lou XE (2010) Dihydroartemisinin improves the efficiency of chemotherapeutics in lung carcinomas In vivo and inhibits murine Lewis lung carcinoma cell line growth In vitro. Cancer Chemother Pharmacol 66(1):21–29
O’Neill PM, Barton VE, Ward SA (2010) The Molecular Mechanism of Action of Artemisinin—the Debate Continues. Molecules 15:1705–1721. doi:10.3390/molecules15031705
Efferth T (2015) Artemisinin–second career as anticancer drug? World J Tradit Chin Med 1(4):2–25
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X et al (2016) Ferroptosis: process and function. Cell Death Differ 23:369–379
Li Q, Weina P, Hickman M (2013) The use of artemisinin compounds as angiogenesis inhibitors to treat cancer, Chap. 7, 10.5772/54109
Dong F, Tian H, Yan S, Li L, Dong X et al (2015) Dihydroartemisinin inhibits endothelial cell proliferation through the suppression of the ERK signaling pathway. Int J Mol Med 35(5):1381–1387. doi:10.3892/ijmm.2015.2140
Zhou Y, Li W, Xiao Y (2016) Profiling of Multiple Targets of Artemisinin Activated by Hemin in Cancer Cell Proteome. ACS Chem Biol. doi:10.1021/acschembio.5b01043
Tran KQ, Tin AS, Firestone GL (2014) Artemisinin triggers a G1 cell cycle arrest of human Ishikawa endometrial cancer cells and inhibits cyclin-dependent kinase-4 promoter activity and expression by disrupting nuclear factor-κB transcriptional signaling. Anticancer Drugs 25(3):270–281. doi:10.1097/CAD.0000000000000054
Tin AS, Sundar SN, Tran KQ, Park AH, Poindexter KM, Firestone GL (2012) Antiproliferative effects of artemisinin on human breast cancer cells requires the downregulated expression of the E2F1 transcription factor and loss of E2F1-target cell cycle genes. Anticancer Drugs 23(4):370–379. doi:10.1097/CAD.0b013e32834f6ea8
Willoughby JA Sr, Sundar SN, Cheung M, Tin AS, Mondiano J, Firestone GL (2009) Artemisinin blocks prostate cancer growth and cell cycle progression by disrupting Sp1 interactions with the Cyclin-dependent Kinase-4 (CDK4) promoter and inhibiting CDK4 gene expression. J Biol Chem 284(4):2203–2213. doi:10.1074/jbc.M804491200
Zhao Y, Jiang W, Li B, Yao Q, Dong J, Cen Y et al (2011) Artesunate enhances radiosensitivity of human non-small cell lung cancer A549 cells via increasing NO production to induce cell cycle arrest at G2/M phase. Int Immunopharmacol 11(12):2039–2046. doi:10.1016/j.intimp.2011.08.017
Chen K, Shou LM, Lin F, Duan WM, Wu MY, Xie X et al (2014) Artesunate induces G2/M cell cycle arrest through autophagy induction in breast cancer cells. Anticancer Drugs 25(6):652–662. doi:10.1097/CAD.0000000000000089
Jiang Z, Chai J, Chuang HH, Li S, Wang T, Cheng Y et al (2012) Artesunate induces G0/G1 cell cycle arrest and iron-mediated mitochondrial apoptosis in A431 human epidermoid carcinoma cells. Anticancer Drugs 23(6):606–613. doi:10.1097/CAD.0b013e328350e8ac
Huang Z, Huang X, Jiang D, Zhang Y, Huang B, Luo G (2016) Dihydroartemisinin inhibits cell proliferation by induced G1 arrest and apoptosis in human nasopharyngealcarcinoma cells. J Can Res Ther 12(1):244–247
Sun H, Meng X, Han J, Zhang Z, Wang B et al (2013) Anti-cancer activity of DHA on gastric cancer–an in vitro and in vivo study. Tumour Biol 34(6):3791–3800. doi:10.1007/s13277-013-0963-0
Chen H, Sun B, Wang S, Pan S, Gao Y, Bai X et al (2010) Growth inhibitory effects of dihydroartemisinin on pancreatic cancer cells: involvement of cell cycle arrest and inactivation of nuclear factor-κB. J Cancer Res Clin Oncol 136(6):897–903
D’Alessandro S, Basilico N, Corbett Y, Scaccabarozzi D, Omodeo-Salè F et al (2011) Hypoxia modulates the effect of dihydroartemisinin on endothelial cells. Biochem Pharmacol 82(5):476–484. doi:10.1016/j.bcp.2011.06.002
Wartenberg M, Wolf S, Budde P, Grünheck F, Acker H, Hescheler J et al (2003) The Antimalaria Agent Artemisinin Exerts Antiangiogenic Effects in Mouse Embryonic Stem Cell-Derived Embryoid Bodies. Lab Invest 83(11):1647–1655
Jia J, Qin Y, Zhang L, Guo C, Wang Y et al (2016) Artemisinin inhibits gallbladder cancer cell lines through triggering cell cycle arrest and apoptosis. Mol Med Rep 13(5):4461–4468. doi:10.3892/mmr.2016.5073
Tong Y, Liu Y, Zheng H, Zheng L, Liu W et al (2016) Artemisinin and its derivatives can significantly inhibit lung tumorigenesis and tumor metastasis through Wnt/β-catenin signaling. Oncotarget 7(21):31413–31428
Eling N, Lukas R, Hazin J, Hamacher-Brady A, Brady NR (2015) Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2:517–532
Button RW, Lin F, Ercolano E, Vincent JH, Hu B, Hanemann CO et al (2014) Artesunate induces necrotic cell death in schwannoma cells. Cell Death Dis 5:e1466. doi:10.1038/cddis.2014.434
Hamacher-Brady A, Stein HA, Turschner S, Toegel I, Mora R, Jennewein N et al (2011) Artesunate activates mitochondrial apoptosis in breast cancer cells via iron catalyzed lysosomal reactive oxygen species production. J Biol Chem 286(8):6587–6601
Steinbrück L, Pereira G, Efferth T (2010) Effects of artesunate on cytokinesis and G2/M cell cycle progression of tumour cells and budding yeast. Cancer Genom Proteom 7(6):337–346
Jeong DE, Song HJ, Lim S, Jeong Lee S, Lim JE et al (2015) Repurposing the anti-malarial drug artesunate as a novel therapeutic agent for metastatic renal cell carcinoma due to its attenuation of tumor growth, metastasis, and angiogenesis. Oncotarget 6(32):33046–33064
Greenshields AL, Shepherd TG, Hoskin DW (2016) Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol Carcinog. doi:10.1002/mc.22474
Jiao Y, Ge C-M, Meng Q-H, Cao J-P, Tong J, Fan S-J (2007) Dihydroartemisinin is an inhibitor of ovarian cancer cell growth. Acta Pharmacol Sin 28(7):1045–1056
Wang Z, Hu W, Zhang J-L, Wu X-H, Zhou H-J (2012) Dihydroartemisinin induces autophagy and inhibits the growth of iron-loaded human myeloid leukemia K562 cells via ROS toxicity. FEBS Open Bio 2:103–112. doi:10.1016/j.fob.2012.05.002
Du XX, Li YJ, Wu CL, Zhou JH, Han Y et al (2013) Initiation of apoptosis, cell cycle arrest and autophagy of esophageal cancer cells by dihydroartemisinin. Biomed Pharmacother 67(5):417–424. doi:10.1016/j.biopha.2013.01.013
Lin R, Zhang Z, Chen L, Zhou Y, Zou P et al (2016) Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett 381(1):165–175. doi:10.1016/j.canlet.2016.07.033
Hui HY, Wu N, Wu M, Liu Y, Xiao SX et al Zhang MF (2016) Dihydroartemisinin suppresses growth of squamous cell carcinoma A431 cells by targeting the Wnt/β-catenin pathway. Anticancer Drugs 27(2):99–105. doi:10.1097/CAD.0000000000000307
Kim SH, Kang SH, Kang BS (2016) Therapeutic effects of dihydroartemisinin and transferrin against glioblastoma. Nutr Res Pract 10(4):393–397
Yang N-D, Tan S-H, Ng S, Shi Y, Zhou J et al (2014) Artesunate Induces Cell Death in Human Cancer Cells via Enhancing Lysosomal Function and Lysosomal Degradation of Ferritin. J Biol Chem 289(48):33425–33441. doi:10.1074/jbc.M114.564567
Mercer AE, Copple IM, Maggs JL, O’Neill PM, Park BK (2011) The role of heme and the mitochondrion in the chemical and molecular mechanisms of mammalian cell death induced by the artemisinin antimalarials. J Biol Chem 286(2):987–996. doi:10.1074/jbc.M110.144188
Du, JH., Zhang, HD., Ma, ZJ., Ji, KM. (2010) Artesunate induces oncosis-like cell death In vitro and has antitumor activity against pancreatic cancer xenografts In vivo. Cancer Chemother Pharma 65:895–902
Liou G-Y, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44(5). doi:10.3109/10715761003667554
Schieber M, Chandel NS (2014) ROS Function in Redox Signaling and Oxidative Stress. Curr Biol 24:R453–R462. doi:10.1016/j.cub.2014.03.034
Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M (2015) Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol 4:184–192
Firestone GL, Sundar SN (2009) Anticancer activities of artemisinin and its bioactive derivatives. Expert Rev Mol Med 11:e32. doi:10.1017/S1462399409001239
Huang C, Ba Q, Yue Q, Li J, Li J, Chu R, Wang H (2013) Artemisinin rewires the protein interaction network in cancer cells: network analysis, pathway identification, and target prediction. Mol BioSyst 9:3091–3100. doi:10.1039/C3MB70342H
He Q, Shi J, Shen XL, An J, Sun H, Wang L et al (2010) Dihydroartemisinin upregulates death receptor 5 expression and cooperates with TRAIL to induce apoptosis in human prostate cancer cells. Cancer Biol Ther 9(10):819–824
Konkimalla VB, Blunder M, Korn B, Soomro SA, Jansen H, Chang W et al (2008) Effect of artemisinins and other endoperoxides on nitric oxide-related signaling pathway in RAW 264.7 mouse macrophage cells. Nitric Oxide 19(2):184–191. doi:10.1016/j.niox.2008.04.008
Lai HC, Singh NP, Sasaki T (2013) Development of artemisinin compounds for cancer treatment. Invest New Drugs 31(1):230–246. doi:10.1007/s10637-012-9873-z
Lai H, Nakase I, Lacoste E, Singh NP, Sasaki T (2009) Artemisinin-transferrin conjugate retards growth of breast tumors in the rat. Anticancer Res 29:3807–3810
Bhadra D, Bhadra S, Jain NK (2005) Pegylated lysine based copolymeric dendritic micelles for solubilization and delivery of artemether. J Pharm Pharmaceut Sci 8(3):467–482
Chen Y, Lin X, Park H, Greever R (2009) Study of artemisinin nanocapsules as anticancer drug delivery systems. Nanomedicine 5(3):316–322. doi:10.1016/j.nano.2008.12.005
Letchmanan K, Shen S-C, Kiong Ng W, Tan RBH (2015) Enhanced dissolution and stability of artemisinin by nano-confinement in ordered mesoporous SBA-15 particles. Microencapsul 32(4):390–400. doi:10.3109/02652048.2015.1035684
Dai L, Wang L, Deng L, Liu J, Lei J, Li D, He J (2014) Novel multiarm polyethylene glycol-dihydroartemisinin conjugates enhancing therapeutic efficacy in non-small-cell lung Cancer. Sci Rep 4:5871. doi:10.1038/srep05871
Lu W-F, Chen S-F, Wen Z-Y, Li Q, Chen J-H (2012) In vitro evaluation of efficacy of dihydroartemisinin-loaded methoxy poly(ethylene glycol)/poly(L-lactic acid) amphiphilic block copolymeric micelles. J Appl Polym Sci. doi:10.1002/APP.38518
Righeschi C, Coronnello M, Mastrantoni A, Isacchi B, Bergonzi MC et al (2014) Strategy to provide a useful solution to effective delivery of dihydroartemisinin: Development, characterization and in vitro studies of liposomal formulations. Colloids Surf B Biointerfaces 116:121–127
Dadgar N, Esfahani MKM, Torabi S, Alavi SE, Akbarzadeh A (2013) Effects of nanoliposomal and pegylated nanoliposomal artemisinin in treatment of breast cancer. Ind J Clin Biochem. doi:10.1007/s12291-013-0389-x
Sun Q, Teong B, Chen I-F, Chang SJ, Gao J, Kuo S-M (2014) Enhanced apoptotic effects of dihydroartemisinin-aggregated gelatin and hyaluronan nanoparticles on human lung cancer cells. J Biomed Mater Res Part B 102B:455–462
Wang Z, Yu Y, Ma J, Zhang H, Zhang H, Wang X et al (2012) LyP-1 modification to enhance delivery of artemisinin or fluorescent probe loaded polymeric micelles to highly metastatic tumor and its lymphatics. Mol Pharm 9:2646–2657. doi:10.1021/mp3002107
Dwivedi A, Mazumder A, du Plessis L, du Preez JL, Haynes RK, du Plessis J (2015) In vitro anti-cancer effects of artemisone nano-vesicular formulations on melanoma cells. Nanomedicine 11(8):2041–2050. doi:10.1016/j.nano.2015.07.010
Liu K, Dai L, Li C, Liu J, Wang L, Lei J (2016) Self-assembled targeted nanoparticles based on transferrin modified eight-arm-polyethylene glycol–dihydroartemisinin conjugate. Sci Rep 6:29461. doi:10.1038/srep29461
Fu J, Zhu Y (2017) Lysosomes activating chain reactions against cancer cells with a pH-switched prodrug/procatalyst co-delivery nanosystem. J Mater Chem B 7(5):996–1004. doi:10.1039/C6TB02820A
Ma W, Xu A, Ying J, Li B, Jin Y (2015) Biodegradable core–shell copolymer-phospholipid nanoparticles for combination chemotherapy: an in vitro study. J Biomed Nanotechnol 11:1193–1200
Li X-Y, Zhao Y, Sun M-G, Shi J-F, Ju R-J, Zhang C-X et al (2014) Multifunctional liposomes loaded with paclitaxel and artemether for treatment of invasive brain glioma. Biomaterials 35:5591–5604
Fröhlich T, Karagöz AC, Reiter C, Tsogoeva SB (2016) Artemisinin-derived dimers: potent antimalarial and anti-cancer agents. J Med Chem. doi:10.1021/acs.jmedchem.5b01380
Alagbala AA, McRiner AJ, Borstnik K, Labonte T, Chang W et al (2006) Biological mechanisms of action of novel C-10 non-acetal trioxane dimers in prostate cancer cell lines. J Med Chem 49:7836–7842
Stockwin LH, Han B, Yu SX, Hollingshead MG, Elsohly MA et al (2009) Artemisinin dimer anticancer activity correlates with heme-catalyzed reactive oxygen species generation and endoplasmic reticulum stress induction. Int J Cancer 125:1266–1275
Posner GH, McRiner AJ, Paik IH, Sur S, Borstnik K et al (2004) Anticancer and antimalarial efficacy and safety of artemisinin-derived trioxane dimers in rodents. J Med Chem 47:1299–1301
Lombard MC, N’Da DD, Breytenbach JC, Kolesnikova NI, Tran Van Ba C, Wein S, Norman J, Denti P, Vial H, Wiesner L (2012) Antimalarial and anticancer activities of artemisinin–quinoline hybrid-dimers and pharmacokinetic properties in mice. Eur J Pharm Sci 47:834–841
Singh NP, Lai HC, Park JS, Gerhardt TE, Kim BJ, Wang S, Sasaki T (2011) Effects of artemisinin dimers on rat breast cancer cells in vitro and in vivo. Anticancer Res 31:4111–4114
Fox JM, Moynihan JR, Mott BT, Mazzone JR, Anders NM et al (2016) Artemisinin-derived dimer ART-838 potently inhibited human acute leukemias, persisted in vivo, and synergized with antileukemic drugs. Oncotarget 7(6):7268–7279
Beekman AC, Barentsen ARW, Woerdenbag HJ, Van Uden W, Pras N, Konings AWT, El-Feraly FS, Galal AM, Wikstrom HV (1997) Stereochemistry-dependent cytotoxicity of some artemisinin derivatives. J Nat Prod 60:325–330
Posner GH, Paik I-H, Sur S, McRiner AJ, Borstnik K, Xie S, Shapiro TA (2003) Orally active, antimalarial, anticancer, artemisinin-derived trioxane dimers with high stability and efficacy. J Med Chem 46:1060–1065
Emens LA, Middleton G (2015) The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res 3(5):436–443. doi:10.1158/2326-6066.CIR-15-0064
Yao W, Wang F, Wang H (2016) Immunomodulation of artemisinin and its derivatives. Sci Bull. doi:10.1007/s11434-016-1105-z
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420(6917):860–867. doi:10.1038/nature01322
Wang X, Lin Y (2008) Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin 29(11):1275–1288
Coffelt SB, Wellenstein MD, de Visser KE (2016) Neutrophils in cancer: neutral no more. Nat Rev Cancer 16:431–446
Hunt S, Yoshida M, Davis CEJ, Greenhill NS, Davis PF (2015) An extract of the medicinal plant Artemisia annua modulates production of inflammatory markers in activated neutrophils. J Inflamm Res 8:9–14
Williams CB, Yeh ES, Soloff AC (2016) Tumor-associated macrophages: unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2:15025–15046. doi:10.1038/npjbcancer.2015.25
Li B, Zhang R, Li J et al (2008) Antimalarial artesunate protects sepsis model mice against heat-killed Escherichia coli challenge by decreasing TLR4, TLR9 mRNA expressions and transcription factor NF-kappa B activation. Int Immunopharmacol 8:379–389
Wang Y, Huang ZQ, Wang CQ et al (2011) Artemisinin inhibits extracellular matrix metalloproteinase inducer (EMMPRIN) and matrix metalloproteinase-9 expression via a protein kinase Cdelta/p38/extracellular signal-regulated kinase pathway in phorbol myristate acetate-induced THP-1 macrophages. Clin Exp Pharmacol Physiol 38:11–18
Yu WY, Kan WJ, Yu PX et al (2012) Anti-inflammatory effect and mechanism of artemisinin and dihydroartemisinin. China J Chin Mater Med 37:2618–2621. (in Chinese)
Wu B, Hu K, Li S et al (2012) Dihydroartiminisin inhibits the growth and metastasis of epithelial ovarian cancer. Oncol Rep 27:101–108
Kitamura T, Qian B-Z, Pollard JW (2015) Immune cell promotion of metastasis. Nat Rev Immunol 15(2):73–86. doi:10.1038/nri3789
Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W et al (2014) Inactivation of the PI3K p110δ breaks regulatory T cell-mediated immune tolerance to cancer. Nature 510(7505):407–411. doi:10.1038/nature13444
Sun XZ (1991) Experimental study on the immunosuppressive effects of qinghaosu and its derivative. Chin J Mod Dev Tradit Med 11:37–38 (in Chinese)
Wang JX, Tang W, Shi LP, Wan J, Zhou R, Ni J et al (2007) Investigation of the immunosuppressive activity of artemether on T-cell activation and proliferation. Br J Pharmacol 150:652–661
Yang DM, Liew FY (1993) Effects of qinghaosu (artemisinin) and its derivatives on experimental cutaneous leishmaniasis. Parasitology 106(Pt 1):7–11
Oleinika K, Nibbs RJ, Graham GJ, Fraser AR (2012) Suppression, subversion and escape: the role of regulatory T cells in cancer progression. Clin Exp Immunol 171:36–45
Langroudi L, Hassan ZM, Ebtekar M, Mahdavi M, Pakravan N, Noori S (2010) A comparison of low-dose cyclophosphamide treatment with artemisinin treatment in reducing the number of regulatory T cells in murine breast cancer model. Int Immunopharmacol 10:1055–1061
Zhang LX, Liu ZN, Ye J, Sha M, Qian H, Bu XH et al (2014) Artesunate exerts an antiimmunosuppressive effect on cervical cancer by inhibiting PGE2 production and Foxp3 expression. Cell Biol Int 38:639–646
Ramacher M, Umansky V, Efferth T (2009) Effect of artesunate on immune cells in ret-transgenic mouse melanoma model. Anti Cancer Drug 20:910–917
Mohamadabadi MA, Hassan ZM, Hosseini AZ, Gholamzad M, Noori S, Mahdavi M et al (2013) Arteether exerts antitumor activity and reduces CD4 + CD25 + FOXP3 + T-reg cells in vivo. Iran J Immunol 10:139–149
Noori S, Hassan ZM (2011) Dihydroartemisinin shift the immune response towards Th1, inhibit the tumor growth in vitro and in vivo. Cell Immunol 271:67–72
Caspi R (2008) Immunotherapy of autoimmunity and cancer: the penalty for success. Nat Rev Immunol 8(12):970–976. doi:10.1038/nri2438
Schieber M, Chandel NS (2014) ROS Function in Redox Signaling and Oxidative Stress. Curr Biol 24(10):R453–R462. doi:10.1016/j.cub.2014.03.034
Chen X, Song M, Zhang B, Zhang Y (2016) Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxidative Medicine and Cellular Longevity. doi:10.1155/2016/1580967
Vieira FGK, Di Pietro PF, Boaventura BCB, Ambrosi C, Rockenbach G et al (2011) Factors associated with oxidative stress in women with breast cancer. Nutr Hosp 26(3):528–536
Mecdad AA, Ahmed MH, ElHalwagy MEA, Afify MMM (2011) A study on oxidative stress biomarkers and immunomodulatory effects of pesticides in pesticide-sprayers. Egyptian. J Forensic Sci 1:93–98. doi:10.1016/j.ejfs.2011.04.012
Brenner DR, Scherer D, Muir K, Schildkraut J, Boffetta P et al (2014) A review of the application of inflammatory biomarkers in epidemiologic. Cancer Res. doi:10.1158/1055-9965.EPI-14-0064
Acknowledgements
The authors are indebted to the Mauritius Research Council for funding drug delivery research and to Bionexx Company (Madagascar) for funding a project related to artemisinin.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors collaborate with Bionexx Company (Madagascar), a supplier of artemisinin, in developing nano-based formulations.
Rights and permissions
About this article

Cite this article
Bhaw-Luximon, A., Jhurry, D. Artemisinin and its derivatives in cancer therapy: status of progress, mechanism of action, and future perspectives. Cancer Chemother Pharmacol 79, 451–466 (2017). https://doi.org/10.1007/s00280-017-3251-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-017-3251-7