
Abstract
Purpose
The use of trastuzumab, a monoclonal antibody targeting the HER2 protein, in combination with 5-fluorouracil/platinum-based chemotherapy improves survival in patients with HER2-positive advanced gastric cancer. In addition, TS-one (S-1)/platinum is also used as a standard of care in Asian countries. However, little is known about the combination of S-1/cisplatin chemotherapy and trastuzumab in patients with HER2-positive advanced gastric/gastroesophageal junction (GEJ) cancer.
Methods
We conducted a single-arm, two-stage, open-label, multicenter phase II study. Trastuzumab was administered intravenously on day 1 of the first cycle at 8 mg/kg and 6 mg/kg on day 1 of subsequent cycles. Cisplatin was administered intravenously at 60 mg/m2 on day 1 of each cycle after trastuzumab. S-1 was administered orally [based on body surface area (BSA)] twice a day for 14 days in a 3-weekly cycle. Patients with BSA of <1.25 received a total of 80 mg of S-1, those with BSA ≥1.5 received 120 mg, and the remaining received 100 mg daily in two divided doses.
Results
All evaluable patients experienced tumor reduction during the trial. The primary end point (overall survival rate) was 59.3 %, with a clinical benefit rate of 66.7 %. Median progression-free survival was 7.4 months; 62.6 % patients were free from disease progression at 6 months. Median overall survival was 14.6 months, and the median time to treatment failure was 6.0 months.
Conclusion
The combination of trastuzumab with S-1 and cisplatin demonstrated good activity, was generally well tolerated, and is a feasible treatment option in the first-line treatment of HER2-positive advanced gastric/GEJ cancers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric cancer is usually diagnosed at an advanced stage. First-line chemotherapy in advanced gastric cancer improves survival and quality of life; furthermore, combination chemotherapy is superior to single-agent chemotherapy [1–7]. HER2 is a key driver of tumorigenesis in gastric cancer [8]. The percentage of gastric cancers demonstrating HER2 overexpression ranges from 6 to 23 % [9–13]. HER2 overexpression is determined by either immunohistochemistry (IHC) or fluorescent in situ hybridization (FISH) [12]. There is a high concordance between IHC and FISH HER2 results from both primary and metastatic sites [12, 14–18].
Trastuzumab is a recombinant monoclonal antibody targeting the extracellular domain of the HER2 protein. Bang et al. [19] showed that the addition of trastuzumab to cisplatin and capecitabine/5-fluorouracil increased the overall survival of patients with advanced HER2-positive gastric/gastroesophageal junction (GEJ) cancers, compared with chemotherapy alone.
S-1 is a novel oral fluoropyrimidine derivative with high oral bioavailability, comprising tegafur, 5-chloro-2,4-dihydropyrimidine, and potassium oxonate [20, 21]. A study conducted in 2010 by Abe et al. [22] suggested that cisplatin combined with TS-one (S-1) may be used as an effective treatment for advanced gastric cancer. Furthermore, studies have also shown that S-1 potentially reduced toxicities compared with 5-fluorouracil [23–25].
In addition, a Japanese phase III trial showed that the combination of S-1 and cisplatin was superior to S-1 alone in advanced gastric cancer [26]. Another phase III trial in non-Asians also studied the combination of cisplatin with either S-1 or infusional fluorouracil. Overall survival was similar, but S-1 was associated with an improved safety profile [27]. These trials showed that the combination of cisplatin and S-1 is a standard chemotherapy for upfront treatment of advanced gastric cancer [26, 28–31].
However, little is known about the combination of S-1/cisplatin chemotherapy and trastuzumab in patients with HER2-positive advanced gastric/GEJ cancer. Hence, the aim of our study was to conduct a single-arm, multicenter phase II study to demonstrate the safety and efficacy of the combination of trastuzumab with S-1 and cisplatin in patients with HER2-positive advanced gastric/GEJ cancers.
Methods
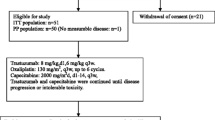
Eligibility
Patients aged ≥21 years with histologically/cytologically confirmed HER2-positive gastric or GEJ cancers were enrolled. HER2 positivity was defined as HER2 IHC 3+, or HER2 IHC2+/FISH positive. A FISH copy number value >2.2 was deemed positive. Post hoc exploratory analysis showed that patients who had IHC 3+ or IHC2+/FISH-positive gastric cancer derived the most benefit. If the patient’s gastric tumor was in situ, obtaining tumor tissue endoscopically was mandatory. If the primary tumor had been resected, collection of further tissue samples was optional.
Patients were required to have measurable disease, European Cooperative Oncology Group (ECOG) performance status ≤2, and life expectancy ≥3 months. They had to be able to consume oral drugs, have normal organ and marrow function, and have a left ventricular ejection fraction (LVEF) of ≥50 %. Normal organ and hematological function were defined as hemoglobin ≥ 8.0 g/dL, leukocytes ≥ 3000/mcL, absolute neutrophil count ≥ 1500/mcL, platelets ≥ 100,000/mcL, total bilirubin ≤ 1.5 times upper limit of normal (ULN), aspartate transaminase (AST)/alanine aminotransferase (ALT)/alkaline phosphatase (ALP) ≤3 times ULN, and creatinine within normal institutional limits/creatinine clearance (using Cockcroft-Gault formula) ≥60 mL/min. In the presence of liver metastasis, AST, ALT, and ALP ≤ 5 times ULN were acceptable.
Patients with prior adjuvant chemotherapy/radiotherapy >180 days before enrollment were eligible, but prior systemic treatment for metastatic disease or any prior use of S-1, cisplatin, or trastuzumab was disallowed. Those with brain metastasis, radiotherapy in the preceding 4 weeks, or any prior malignancy (except basal cell carcinoma and pre-invasive cervical cancer) diagnosed within the last 5 years were ineligible.
Also excluded were patients with serious complications/uncontrolled intercurrent illness that limited compliance with study requirements, those on other investigational agents, human immunodeficiency virus-positive patients, pregnant or lactating patients, and those with reproductive potential not amenable to implementing adequate contraceptive measures. Patients could not have active gastrointestinal bleeding requiring repeated transfusions.
Approval was obtained from the institutional review board of participating centers, and written consent was obtained from all patients.
Study design and sample size
We conducted a single-arm, two-stage, open-label, multicenter phase II study. Based on a sample size of 25 patients, the study was designed to distinguish a favorable overall response rate (ORR) of 55 % from a null rate of 30 % at 5 % significance level and 80 % power. Assuming an approximate 20 % loss to follow-up, the target size for enrollment was 30 patients.
Nine patients were used to evaluate the feasibility of the regimen in the first stage of the trial. An interim analysis based on these patients was performed, the regimen was deemed feasible, and the trial proceeded with second-stage patient accrual.
To speed up accrual, the trial changed from a single-center trial in the first stage to a multicenter trial with four recruitment sites in the second stage. Thirty patients were recruited in total, received trial stipulated treatment drugs, and were included in analysis.
Treatment plan
Chemotherapy was administered in a 3-weekly cycle. Trastuzumab was given intravenously at 8 mg/kg on day 1 of the first cycle, followed by 6 mg/kg on day 1 of subsequent cycles. Cisplatin 60 mg/m2 was intravenously administered on day 1 of each cycle after trastuzumab. Hydration and antiemetic medication prior to and following cisplatin administration were administered according to institutional practice.
S-1 was administered orally twice a day for 14 days, followed by a rest period of 7 days. The initial dose of S-1 was determined based on body surface area (BSA). Patients with BSA < 1.25 received a total of 80 mg of S-1, those with BSA ≥ 1.5 received 120 mg, while all others received 100 mg daily in two divided doses.
Study treatment was continued until disease progression, unacceptable adverse events (AE), treatment delay of more than 3 weeks as a result of an AE, withdrawal of consent, or investigator’s decision. Cisplatin could be discontinued after six cycles at investigator’s discretion.
Dose reduction was permitted level by level with the lowest dose of S-1 and cisplatin set to 80 mg/day and 40 mg/m2, respectively. Dose escalation was not permitted. Dose reductions were sequential for toxicity; cisplatin was reduced to 50 mg/m2 and subsequently to 40 mg/m2. S-1 dose was reduced by 20 mg/day for each dose reduction.
Cardiac assessment by MUGA Scan/2D-Echocardiography was performed prior to enrollment and every 3 months subsequently. Trastuzumab could be interrupted/discontinued for infusional reactions. Trastuzumab was withheld for at least 4 weeks and LVEF reassessed if there was a >16 % absolute decrease from baseline value or if it fell below the institution’s normal limits and was at least a 10 % absolute decrease from pre-treatment value. Trastuzumab could be resumed if LVEF returned to normal limits and the absolute decrease from baseline was <15 % within 4–8 weeks. Trastuzumab was permanently discontinued if there was persistent (>8 weeks) LVEF decline or if its administration had to be suspended on more than three occasions for cardiomyopathy.
Evaluations
The primary end point was overall response rate (ORR). Secondary end points included progression-free survival (PFS), overall survival (OS), time to treatment failure (TTF), clinical benefit rate (CBR), duration of response (DR), and safety of the treatment regimen. Response and progression were evaluated using RECIST version 1.0 [25].
Patients were evaluated every 6 weeks with computed tomography/magnetic resonance imaging scans. Confirmatory scans were obtained at least 4 weeks following initial documentation of objective response. Patients who ended study treatment without progressive disease (PD) were followed for tumor response every 6 weeks from the end of study until PD or the initiation of any post-study anti-tumor therapy, whichever was earlier. All patients were followed up for 24 months after the last patient had initiated study treatment or until death, whichever occurred first. Survival status was assessed every 24 weeks over the phone from the end of study treatment.
OS was defined as time from registration to date of death. PFS was defined as time from registration until objective tumor progression or death from any cause, whichever occurred first. TTF was a composite end point measuring time from registration until treatment discontinuation. DR was defined as time from first assessment of CR (complete response) or PR (partial response) until the first date of PD or death within 60 days of the last tumor assessment or registration, whichever occurred first. Toxicities were graded according to the NCI CTCAE version 3.0.
Statistical analysis
All patients who received study-mandated treatment were included in the safety and efficacy analysis. Response rates were also assessed among evaluable patients, defined as those with measurable disease, had received ≥1 cycle of therapy, and had their disease re-evaluated.
Descriptive statistics were used to summarize patient demographics, baseline clinical characteristics, treatment modifications, and toxicities. Relative dose intensity (RDI) of each drug was estimated as the proportion of actual total dose to planned total dose received by patient until drug discontinuation. ORR and CBR were estimated using exact method [26]. Each survival distribution was estimated using Kaplan–Meier method. Exploratory association analysis of end points with demographics and clinical characteristics was assessed based on odds ratios for categorical outcomes and hazard ratios for survival outcomes. Statistical analyses were performed using SAS version 9.3 (SAS Institute Inc., Cary, NC).
Results
Patient characteristics
Between April 2010 and April 2013, a total of 30 patients were recruited, of whom three discontinued treatment.
Of the 30 recruited patients, one discontinued treatment during cycle 1 due to serious infusion reaction to trastuzumab, one refused treatment in cycle 1 after trastuzumab infusion, and another demised before the end of cycle 2 from treatment-related neutropenic sepsis. All three patients did not have their disease re-evaluated on treatment discontinuation. Twenty-four patients discontinued treatment as of September 30, 2013; reasons for discontinuation are listed in “Appendix 1.”
Median age at diagnosis was 61.9 years (range 43.5–79.8 years) (Table 1). Majority were males (73 %), had primary tumor in the stomach (97 %), and had no prior resection of primary tumor (90 %). All had good performance status (ECOG 0 or 1). Majority (73 %) of patients’ HER2 status was based on IHC3+. Sixty percent were histologically diagnosed with intestinal-type gastric cancer, and most were metastatic (87 %).
Treatment compliance
Four patients required dose reduction, 18 dose delay, and two dose omission of trastuzumab (“Appendix 2”). Most dose delays were hematology related. For cisplatin, 13 patients required dose reduction, 22 dose delay, and 19 dose omission. For S-1, 14 patients required dose reduction, 20 dose delay, and five dose omission.
Relative dose intensities were 99.8 % for trastuzumab, 94.0 % for cisplatin, and 89.8 % for S-1 (Table 2). Half of the patients received the full dose for each drug.
Efficacy
Intention to treat analysis of the 30 patients showed one CR and 15 PR (Table 3). The ORR was 53.3 % (95 % CI 34.3–71.7 %). Two additional patients had SD, leading to a CBR of 60.0 % (95 % CI 40.6–77.3 %). The ORR and CBR among the 27 evaluable patients were 59.3 % (95 % CI 38.8–77.6 %) and 66.7 % (95 % CI 46.0–83.5 %), respectively. All evaluable patients experienced tumor reduction during the trial (Fig. 1).

Waterfall plot (among evaluable patients)
With a median follow-up duration of 8.5 months, the median PFS was 7.4 months (95 % CI 5.5–11.0 months) with 62.6 % of patients free from disease progression at the 6-month mark (Table 4). Eighteen patients were still alive at the time of analysis. Median OS was 14.6 months (Fig. 2), and median TTF was 6 months (95 % CI 4.8–7.6 months). Among the 16 patients who responded to the treatment, the median DR was 7.3 months.

Overall survival and progression-free survival curves
None of the demographics and baseline clinical characteristics was significantly associated with ORR, CBR, PFS, or OS, including tumor HER2 status, histology, or primary tumor site.
Toxicities
All patients had at least one AE or serious adverse event (SAE). The commonest non-SAEs (any grade) included fatigue/lethargy (16 patients; 53.3 %), anorexia (53.3 %), diarrhea (46.7 %), and nausea (46.7 %) (Table 5). Majority (70 %) had at least one incidence of grade 3/4 AE. Common grade 3/4 non-SAE included neutropenia (23.3 %), low absolute neutrophil count (20.0 %), and mucositis/stomatitis (13.3 %).
Two patients suffered cardiac AEs; none led to permanent treatment discontinuation/death (Table 6). Half of the patients experienced a decline in LVEF from baseline during treatment; none suffered a LVEF drop of ≥50 % from baseline. Of these 15 patients, nine had a drop in LVEF of <10 % from baseline, while the remaining six patients suffered a >10 % (but <50 %) LVEF drop from their baseline readings. There were no incidences of cardiac failure or cardiac toxicity-related deaths.
Six patients discontinued study drugs as a result of AE or at investigator’s decision. A total of 30 SAEs were reported. Two of them were fatal—one deemed a study drug-related grade 5 toxicity, while the other developed electrolyte and sugar disturbances and turned hypotensive.
Discussion
In our multicenter phase II single-arm study, high efficacy was observed with an ORR of 53.3 % in all patients and close to 60 % in the 27 evaluable patients. CBR was 60.0 and 66.7 %, respectively. All evaluable patients experienced tumor shrinkage during the course of the trial. With a median follow-up duration of 8.5 months, the median PFS and OS were numerically comparable to that seen in the ToGA study. There did not appear to be any difference in outcome between those with HER2 IHC 3+ and those with IHC2 2+/FISH+ cancers.
Similar to our study, Kurokawa et al. [31] reported a phase II Japanese trial combining trastuzumab with cisplatin and S-1 in HER2-positive advanced gastric cancers. They reported a response rate of 68 % (95 % CI 54–80 %) and a disease control rate of 94 % (95 % CI 84–99 %). Median OS, PFS, and TTF were estimated at 16, 7.8, and 5.7 months, respectively. Grade 3/4 AEs include neutropenia (36 %), anorexia (23 %), and anemia (15 %).
Chemotherapy was administered in a similar schedule in both studies, and both efficacy and toxicity end points were comparable. There was one death from neutropenic sepsis/infection attributable to treatment. Apart from that, the AEs and SAEs reported were expected, with no new safety concerns. There were no reported cardiac failure or cardiac toxicity-related deaths; of those who experienced LVEF drop, majority (60 %) had a <10 % drop from baseline.
In conclusion, the combination of trastuzumab with S-1 and cisplatin administered in a 3-weekly cycle demonstrated comparable efficacy to trastuzumab/cisplatin/capecitabine or fluorouracil and was generally well tolerated. This combination is an alternative option in the treatment of advanced HER2-positive gastric/GEJ cancer.
References
Cervantes A, Roda D, Tarazona N, Rosello S, Perez-Fidalgo JA (2013) Current questions for the treatment of advanced gastric cancer. Cancer Treat Rev 39(1):60–67
Glimelius B, Ekstrom K, Hoffman K, Graf W, Sjoden PO, Haglund U et al (1997) Randomized comparison between chemotherapy plus best supportive care with best supportive care in advanced gastric cancer. Ann Oncol 8(2):163–168
Wagner AD, Grothe W, Haerting J, Kleber G, Grothey A, Fleig WE (2006) Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate data. J Clin Oncol 24(18):2903–2909
Wagner AD, Unverzagt S, Grothe W, Kleber G, Grothey A, Haerting J et al (2010) Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev. doi:10.1002/14651858.CD004064.pub3
Cunningham D, Okines AF, Ashley S (2008) Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 358(1):36–46
Van Cutsem E, Moiseyenko VM, Tjulandin S, Majlis A, Costenla M, Boni C et al (2006) Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 24(31):4991–4997
Koizumi W, Narahara H, Hara T, Takagane A, Akiya T, Takagi M, Miyashita K, Nishizaki T, Kobayashi O, Takiyama W, Toh Y, Nagaie T, Takagi S, Yamamura Y, Yanaoka K, Orita H, Takeuchi M (2008) S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol 9(3):215–221
Gunturu KS, Woo Y, Beaubier N, Remotti HE, Saif MW (2013) Gastric cancer and trastuzumab: first biologic therapy in gastric cancer. Ther Adv Med Oncol 5(2):143–151
Begnami MD, Fukuda E, Fregnani JH, Nonogaki S, Montagnini AL, da Costa WL Jr et al (2011) Prognostic implications of altered human epidermal growth factor receptors (HERs) in gastric carcinomas: HER2 and HER3 are predictors of poor outcome. J Clin Oncol 29(22):3030–3036
Grabsch H, Sivakumar S, Gray S, Gabbert HE, Muller W (2010) HER2 expression in gastric cancer: rare, heterogeneous and of no prognostic value—conclusions from 924 cases of two independent series. Cell Oncol 32(1–2):57–65
Gravalos C, Jimeno A (2008) HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Ann Oncol 19(9):1523–1529
Hofmann M, Stoss O, Shi D, Buttner R, van de Vijver M, Kim W et al (2008) Assessment of a HER2 scoring system for gastric cancer: results from a validation study. Histopathology 52(7):797–805
Hsu JT, Chen TC, Tseng JH, Chiu CT, Liu KH, Yen CN et al (2011) Impact of HER-2 overexpression/amplification on the prognosis of gastric cancer patients undergoing resection: a single-center study of 1,036 patients. Oncologist 16(12):1706–1713
Bozzetti C, Negri FV, Lagrasta CA, Crafa P, Bassano C, Tamagnini I et al (2011) Comparison of HER2 status in primary and paired metastatic sites of gastric carcinoma. Br J Cancer 104(9):1372–1376
Marx AH, Tharun L, Muth J, Dancau AM, Simon R, Yakebas E et al (2009) HER-2 amplification is highly homogenous in gastric cancer. Hum Pathol 40(6):769–777
Reichelt U, Duesedau P, Tsourlakis MC, Quaas A, Link BC, Schurr PG et al (2007) Frequent homogeneous HER-2 amplification in primary and metastatic adenocarcinoma of the esophagus. Mod Pathol 20(1):120–129
Ruschoff J, Dietel M, Baretton G, Arbogast S, Walch A, Monges G et al (2010) HER2 diagnostics in gastric cancer-guideline validation and development of standardized immunohistochemical testing. Virchows Arch 457(3):299–307
Tanner M, Hollmen M, Junttila TT, Kapanen AI, Tommola S, Soini Y et al (2005) Amplification of HER-2 in gastric carcinoma: association with Topoisomerase IIalpha gene amplification, intestinal type, poor prognosis and sensitivity to trastuzumab. Ann Oncol 16(2):273–278
Bang YJ, Van Custem E, Feyereislova A, Chung HC, Shen L, Sawaki A et al (2010) Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376(9742):687–697
Chu QS, Hammond LA, Schwartz G, Ochoa L, Rha SY, Denis L et al (2004) Phase I and pharmacokinetic study of the oral fluoropyrimidine S-1 on a once-daily-for-28-day schedule in patients with advanced malignancies. Clin Cancer Res 10(15):4913–4921
Shirasaka T, Shimamato Y, Oshimo H, Yamagochi M, Kato T, Yonekura K et al (1996) Development of a novel form of an oral 5-fluorouracil derivative (S-1) directed to the potentiation of the tumor selective cytotoxicity of 5-fluorouracil by two biochemical modulators. Anticancer Drugs 7(5):548–557
Abe S, Tsuji Y, Tsushima T, Kogawa T, Abe M, Onodera Y et al (2010) Efficacy and feasibility of combination chemotherapy with S-1 and cisplatin (2 weeks regimen) for advanced gastric cancer. Jpn J Clin Oncol 40(4):302–306
Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 92(3):205–216
Clopper C, Pearson S (1934) The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika 26:404–413
Nagashima F, Ohtsu A, Yoshida S, Ito K (2005) Japanese nationwide post-marketing survey of S-1 in patients with advanced gastric cancer. Gastric Cancer 8(1):6–11
Koizumi W, Narahara H, Hara T, Takagane A, Akiya T, Takagi M et al (2008) S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol 9(3):215–221
Ajani JA, Rodriguez W, Bodoky G, Moiseyenko V, Lichinitser M, Gorbunova V et al (2010) Multicenter phase III comparison of cisplatin/S-1 with cisplatin/infusional fluorouracil in advanced gastric or gastroesophageal adenocarcinoma study: the FLAGS trial. J Clin Oncol 28(9):1547–1553
Yamanaka T, Matsumoto S, Teramukai S, Ishiwata R, Nagai Y, Fukushima M (2008) Safety evaluation of oral fluoropyrimidine S-1 for short- and long-term delivery in advanced gastric cancer: analysis of 3,758 patients. Cancer Chemother Pharmacol 61(2):335–343
Boku N, Yamamoto S, Fukuda H, Shirao KT, Sawaki A et al (2009) Fluorouracil versus combination of irinotecan plus cisplatin versus S-1 in metastatic gastric cancer: a randomised phase 3 study. Lancet Oncol 10(11):1063–1069
Sato Y, Kondo H, Honda K, Takahari D, Sumiyoshi T, Tsuiji Y et al (2005) A phase I/II study of S-1 plus cisplatin in patients with advanced gastric cancer: 2-week S-1 administration regimen. Int J Clin Oncol 10(1):40–44
Kurokawa Y, Sugimoto N, Miwa H, Tsuda M, Nishina S, Okuda H et al (2014) Phase II study of trastuzumab in combination with S-1 plus cisplatin in HER2-positive gastric cancer (HERBIS-1). Br J Cancer 110(5):1163–1168
Acknowledgments
We are grateful to Taiho Pharmaceutical Co. for supporting this trial with a study Grant and to Professor Atsushi Ohtsu who was an advisor. We would like to thank Ms Yeo Ai Ling for her assistance in editing this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Co-author, Dr. Choo Su-Pin, received a travel Grant from Taiho (which also provided the study Grant). Co-author, Dr. Yasuhide Yamada, received an honoraria and research Grant from Taiho.
Rights and permissions
About this article

Cite this article
Chua, C., Tan, I.B., Yamada, Y. et al. Phase II study of trastuzumab in combination with S-1 and cisplatin in the first-line treatment of human epidermal growth factor receptor HER2-positive advanced gastric cancer. Cancer Chemother Pharmacol 76, 397–408 (2015). https://doi.org/10.1007/s00280-015-2811-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-015-2811-y