
Abstract
Purpose
Fluoropyrimidines and oxaliplatin have demonstrated some efficacy against pancreatic adenocarcinoma, but survival remains brief. Sorafenib is an oral multikinase inhibitor which we sought to combine with a unique capecitabine and oxaliplatin regimen for pancreatic adenocarcinoma.
Methods
We performed a multicenter phase II study of sorafenib 200 mg orally twice daily along with oxaliplatin 85 mg/m2 IV on days 1 and 15, followed by capecitabine 2250 mg/m2 orally every 8 h for six doses starting on days 1 and 15 of a 28-day cycle in patients who had no more than one previous chemotherapy regimen for their pancreatic adenocarcinoma. The primary objective was response rate; secondary objectives were progression-free survival (PFS), overall survival (OS), and safety.
Results
Twenty-four patients were enrolled; median age was 63 years (range 48–83). The most common related toxicities were fatigue, neuropathy, anemia, thrombocytopenia, diarrhea, nausea, leukopenia, and hand-foot syndrome. Grade 3 hand-foot syndrome was rare (4 %). Other grade 4 toxicities included abdominal pain (8 %), pulmonary embolism (4 %), and anemia (4 %). Three partial responses were seen (13 %), and 11 patients had stable disease (46 %) as their best response. Median PFS was 6.0 months (range 1.5–13 months). Median OS was 8.1 months (range 1.5–13.6 months).
Conclusions
Sorafenib, oxaliplatin, and capecitabine produced partial responses in patients with advanced pancreatic cancer including previously treated patients and demonstrated a PFS of 6 months with few grade 3/4 toxicities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In 2015, an estimated 48,960 people will develop exocrine pancreatic cancer in the USA and 40,560 will die from this disease [1]. This high burden of mortality reflects the disease’s aggressive character, and most patients present with advanced disease. Chemotherapy provides a modest survival benefit. For example, the MPACT study showed an overall survival of 8.5 months for patients receiving a combination of gemcitabine and nab-paclitaxel [2]. A combination of 5-fluorouracil (5-FU), leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX) has demonstrated a significant improvement over gemcitabine in patients with a good performance status, with a median survival of 11.1 months in patients treated with FOLFIRINOX versus 6.8 months with gemcitabine. However, this regimen is associated with significant toxicity, particularly myelosuppression [3]. Given the modest efficacy and significant toxicities associated with current regimens, further investigation of new regimens is required.
A novel chemotherapy regimen meant to replicate modified FOLFOX6 (oxaliplatin 85 mg/m2 and leucovorin 400 mg/m2 on day 1 followed by a 5-FU bolus 400 mg/m2 and a 46-h infusion of 3000 mg/m2 every 2 weeks) has been developed [4]. In this regimen, high-dose capecitabine given over 2 days has replaced the continuous infusion of 5-FU. This contrasts with the usual 7- or 14-day course of capecitabine for other regimens [5, 6]. This regimen has been called oral FOLFOX6 or 2-day oxaliplatin–capecitabine (2DOC). A phase I evaluation of 2DOC was performed with the recommended phase two doses being oxaliplatin 85 mg/m2 intravenously with capecitabine 2250 mg/m2 orally every 8 h for six doses (2 days), with both drugs given on days 1 and 14 of a 28-day cycle [7]. Of note in that study, there was no evidence of saturation of capecitabine metabolism. Even at near twice standard doses of capecitabine, conversion to 5-FU followed a predictable time course and the AUCs and C max of capecitabine, 5′-DFCR, 5′-DFUR, and 5-FU were all within published ranges [7].
With the phase 1 study of the 2DOC regimen, promising responses were seen in patients with pancreatic adenocarcinoma. For example, of six patients treated, there was one minor response (21 % shrinkage, with reduction of CA 19-9 from 14,370 to 887 units/mL on therapy) and one patient with stable disease. Notably, both of these patients had gemcitabine refractory disease [7].
Sorafenib (Nexavar, BAY 43–9006) is an oral multikinase inhibitor with effects on tumor proliferation and tumor angiogenesis. Sorafenib has activity against c-RAF, BRAF, VEGFR-2, PDGFR-β, c-kit, and FLT3 [8]. Pancreatic cancer contains targets where sorafenib could be considered an attractive targeted agent. For example, 70–90 % of pancreas cancers have Ras activation [9]. Moreover, sorafenib has shown efficacy in human and cell line pancreas cancer models [10, 11]. However, palmar-plantar erythrodysesthesia remains one of the major dose-limiting toxicities of sorafenib [12].
In addition to rare events of palmar-plantar erythrodysesthesia seen on the 2DOC phase I study, a phase II study comparing 2DOC to traditional dosing of capecitabine and oxaliplatin in metastatic colorectal cancer found similar overall response rates, time to progression, and overall survival, but significantly decreased hand-foot syndrome, diarrhea, and mucositis in the 2DOC group [13]. The lack of hand-foot syndrome with 2DOC made it an ideal regimen for combination with tyrosine kinase inhibitors (TKIs) such as sorafenib, as there are few expected overlapping toxicities.
Our recently conducted phase I study combining 2DOC with sorafenib in pancreaticobiliary cancer patients showed a reasonable safety profile with a recommended phase II dose of sorafenib 200 mg BID [14]. Most notably, there did not appear to be synergistic palmar-plantar erythrodysesthesia between 2DOC and sorafenib as the rate and severity of palmar-plantar erythrodysesthesia were similar to that expected from single-agent sorafenib. Moreover, that study did show a partial response in one of the six patients with pancreatic cancer and stable disease in another. Based on the phase I results showing the safety of a combination of 2DOC and sorafenib, we sought primarily to assess the clinical response rate of patients with advanced pancreas cancer.
Patient and methods
This phase II study was performed in the outpatient setting at six institutions within the Wisconsin Oncology Network (WON). WON is a regional clinical research network of academic and community health centers throughout Wisconsin, South Dakota, and northern Illinois. It was approved by the institutional review board at the University of Wisconsin Carbone Cancer Center and all participating WON centers. Informed consent was obtained from all patients prior to their participation in the study.
Patient eligibility
Patients with histologically or cytologically confirmed locally advanced or metastatic adenocarcinoma of the pancreas who had not received more than one previous systemic treatment were included. Patients must have demonstrated at least one measurable lesion as defined by RECIST 1.1 [15]. Other eligibility criteria included age of at least 18 years, ECOG performance status of 0–2, less than grade 2 neuropathy, and adequate organ and marrow function defined as leukocytes greater than 3000/µL, absolute neutrophil count greater than 1500/µL, platelets greater than 100,000/µL, total bilirubin <2.5 times the institutional upper limit of normal, AST(SGOT) and ALT(SGPT) <5 times the institutional upper limit of normal, and creatinine clearance greater than 50 mL/min as calculated by the Cockroft–Gault formula. Women of childbearing potential and men agreed to use adequate contraception throughout the duration of the study. Other important exclusion criteria included concomitant radiation therapy or systemic cancer therapies; known brain metastases; history of allergy to platinum compounds, capecitabine, sorafenib, or antiemetics; uncontrolled intercurrent illness; pregnant or nursing women; prior systemic therapy, radiotherapy, major surgery, open biopsy, or significant traumatic injury within 4 weeks of first study drug; inability to swallow whole pills; second malignancy within the past 3 years (excluding nonmelanoma skin cancer and in situ cancers) that had not been treated with curative intent or with current evidence of disease; and known gastrointestinal malabsorption syndromes.
Treatment and dose modifications
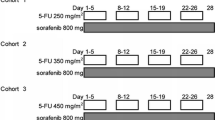
On days 1 and 15 of a 28-day cycle, each patient received oxaliplatin 85 mg/m2 administered as a 2-h intravenous infusion plus capecitabine 2250 mg/m2 orally every 8 h for a total of six doses. Additionally, oral sorafenib at a dose of 200 mg BID was administered each day of the 28-day cycle (Fig. 1). In the absence of treatment delays due to toxicity, treatment continued until one of the following: disease progression, comorbid illness preventing further administration of treatment, unacceptable toxicity, patient withdrawal from the study, or general or specific changes in the patient’s condition rendering the patient unacceptable for further treatment in the judgment of the investigator including the patient becoming eligible for an attempt at surgical resection.

Treatment schema
Before beginning a subsequent cycle of therapy, patients were required to have an ANC greater than 1000/µL and platelet count over 75,000/µL. All toxicities were graded according to the NCI Common Terminology Criteria for Adverse Events, Version 3.0 [16]. Prior to receiving the next cycle, patients needed to have grade 1 or less stomatitis/pharyngitis, diarrhea, or skin toxicity, as well as grade 2 or less paresthesias/dysesthesias. Dose modification of oxaliplatin was made for grade 3 or 4 peripheral neuropathy of greater than 7-day duration. For the first incidence of grade 3 or 4 neuropathy, the dose was reduced to 65 mg/m2 or by 25 % if the starting dose was <85 mg/m2. If the neuropathy persisted after the dose-reduced cycle, oxaliplatin was discontinued. For all other grade 3 or greater toxicities, treatment was held until the toxicity was grade 1 or less. For toxicities considered to be related to sorafenib, dosing was first reduced to 200 mg daily and discontinued if prior dosing was 200 mg daily. All other medications were reduced by 25 % for the next cycle. The number of dose modifications allowed was at the discretion of the treating physician.
Study end points
The primary end point was to assess the response rate of the combination of 2DOC and sorafenib in patients with locally advanced or metastatic pancreatic cancer by RECIST 1.1 [15]. The secondary end points were progression-free survival, overall survival, and safety.
Statistical methods
Demographic variables, toxicities, and responses were summarized in terms of frequencies and percentages. Each patient was assigned one of the following categories: complete response, partial response, stable disease, progressive disease, early death from malignant disease, early death from toxicity, early death because of other cause, or unknown. Patients not in the first two response categories were considered as failing to respond to the proposed treatment combination. The overall response rate was calculated as the proportion of patients with a complete or partial response out of all patients enrolled. The Wilson score method was used to construct the 95 % confidence interval of the response rate. Toxicities (categorized as at least possibly treatment related) were summarized in tabular format by type and severity. The Kaplan–Meier method was used to estimate duration of response, progression-free survival, and overall survival. Progression-free survival was defined as the number of months from the day of first study drug administration to the day the patient experienced an event of disease progression or death, whichever occurred first. If a patient had not experienced an event of disease progression (or death) at the end of the follow-up period, then the patient’s data were censored at the date of the last available evaluation. Overall survival was defined as the number of months from the day of first study drug administration to the day the patient died from any cause. If a patient was still alive at the time of analysis, then the patient’s data were censored at the date of the last available evaluation. Progression-free survival and overall survival were displayed in graphical format using Kaplan–Meier curves. Statistical analysis was conducted using SAS software version 9.3 (SAS Institute Inc., Cary, NC).
Results
Patient characteristics
A total of 24 patients enrolled, which included 13 men and 11 women (Table 1). The median age was 63 years (range 48–83 years). Fifteen patients (62 %) had received prior gemcitabine-containing therapy. Two patients (8 %) had prior pancreatectomies, while three patients (13 %) had previous pancreatic radiotherapy.
Safety and tolerability
All 24 patients were evaluated for toxicity with results summarized for toxicities at least possibly related to treatment in Table 2. The most common toxicities considered to be at least possibly related to study treatment were fatigue (75 %), neuropathy (58 %), anemia (58 %), low platelets (58 %), diarrhea (46 %), nausea (42 %), leukopenia (42 %), and hand-foot syndrome (33 %); most were grade 1–2. Grade 3 hand-foot syndrome was rare (one patient, 4 %). Four (17 %) cases of grade 4 toxicities were observed, including one (4 %) case of pulmonary embolism, two (8 %) cases of abdominal pain, and one (4 %) case of anemia; 33 % of the toxicities occurred during cycle 1, 12 % during cycle 2, 19 % during cycle 3, and 36 % in cycle 4 or afterwards. The most common reason for discontinuation was disease progression (54 %).
Efficacy
Of the 24 patients enrolled, 19 were evaluable for response. Three (13 %) partial responses were seen, and an additional eleven patients (46 %) had stable disease (Table 3). The overall response rate was 13 % (95 % CI 4–31 %). Of note, two of the three patients with a partial response had received prior treatment with gemcitabine. The median duration of the partial response was 7.5 months, while the median duration of stable disease was 4.3 months. Median progression-free survival was 6.0 months (95 % CI 2.5–9.6) (Fig. 2), while the median overall survival was 8.1 months (95 % CI 3.5–10.9) (Fig. 3).

Kaplan–Meier curve (95 % CI) for progression-free survival

Kaplan–Meier curve (95 % CI) for overall survival
Discussion
In this phase II trial, a combination of 2 days of high-dose capecitabine and oxaliplatin (“2DOC”) and sorafenib showed favorable activity in advanced or metastatic pancreatic cancer with a response rate of 13 % and a disease control rate (PR + SD) of 59 %. The results are particularly notable because the majority of patients were already refractory to gemcitabine. This study demonstrates that even after failure of front-line gemcitabine regimens, fluoropyrimidine-/oxaliplatin-based regimens are still effective. In addition to reasonable response rates, the median duration of partial responses was 7.5 months while the median duration of stable disease was 4.3 months; this compares more favorably than other capecitabine and oxaliplatin regimens for pancreas cancer where the median time to progression was approximately 4 months [17, 18].
When this trial was conceived, neither gemcitabine/nab-paclitaxel nor FOLFIRINOX had become standard of care for front-line therapy in advanced pancreatic cancer. Although FOLFIRINOX has provided relatively excellent response rates and improvements to overall survival in metastatic pancreatic adenocarcinoma, these come at the cost of increased toxicity [3]. Due to concerns about this toxicity, many oncologists prefer gemcitabine/nab-paclitaxel as their initial metastatic pancreas cancer regimen [19]. However, how to optimally sequence in oxaliplatin regimens after gemcitabine/nab-paclitaxel is unknown. Some studies have shown relatively poor activity of FOLFOX in pretreated metastatic pancreatic cancer patients [20]. However, other studies have shown modest response rates for FOLFOX-like regimens [17, 21]. For example, Berk et al. showed response rates in the second-line setting of 18 and 17 % for XELOX and FOLFOX4, respectively [18]. The results of our study further support the use of capecitabine/oxaliplatin as second-line regimen in patients who received a gemcitabine-based regimen for their initial treatment.
Also notable for this combination was that very little hand-foot syndrome was seen, which has been the major difficulty in combining capecitabine and sorafenib. For example, rates of up to 90 % for any grade hand-foot syndrome and up to 44 % for grade 3 hand-foot syndrome have been reported with a combination of standard schedule capecitabine and sorafenib [22]. In contrast, in our study, we saw only one(4 %) incident of grade 3 hand-foot syndrome. We believe that this is because of the alternate capecitabine schedule used in our study. Prior work has shown that by giving the capecitabine over 2 days, rather than 14, one still achieves equivalent pharmacokinetic parameters to infusional 5-FU with FOLFOX6, including a lack of saturation of capecitabine metabolism with the higher doses of capecitabine used in the 2DOC regimen [7].
In addition to low rates of hand-foot syndrome, 2DOC/sorafenib was otherwise well tolerated. The most common adverse events were expected from this combination of chemotherapy. Only four patients (17 %) experienced grade 4 toxicities considered to be related to study treatment including one (4 %) case of pulmonary embolism, two (8 %) cases of abdominal pain, and one (4 %) case of anemia which was similar to 2DOC alone in other studies [13]. Furthermore, there were only four (17 %) instances of grade 3 neutropenia and no grade 4 neutropenia seen. This compares favorably to the 48 % rate of grade 3/4 neutropenia seen in mFOLFOX6 plus sorafenib given in first line to colorectal cancer patients [23]. Given that bolus 5-FU is most strongly associated with neutropenia [24], the lack of neutropenia seen in the 2DOC regimen is likely accounted for the lack of bolus 5-FU.
Other common related grade 3/4 toxicities were fatigue (17 %), abdominal pain (17 %), diarrhea (13 %), neuropathy (13 %), and anemia (13 %). Fatigue and diarrhea are associated with the use of sorafenib and may have contributed to the greater rates of grade 3/4 fatigue and anemia seen in this trial compared to others where 2DOC was used alone [13]. Similarly, when sorafenib has been combined with mFOLFOX6, significant increases in grade 3/4 diarrhea were observed [23]. Interestingly, our study shows greater rates of 3/4 anemia than observed in other studies combining sorafenib with capecitabine [22] or mFOLFOX6 [23]. This may be attributable to the treatment population in this study as 62 % had prior chemotherapy and 13 % had prior radiation.
In addition to showing safety and efficacy, novel chemotherapeutic regimens should ideally translate to clinical scenarios encountered at both academic and community centers. A significant strength of this study was that it was conducted through the Wisconsin Oncology Network taking place at six different cancer centers, most of which are community based. Moreover, this regimen is mostly oral with no need for continuous infusion or central intravenous access, potentially making it easier to administer and more convenient for patients. Thus, the combination of 2DOC and sorafenib can feasibly be given to patients in both academic and community settings.
Although sorafenib has shown promise in human pancreatic cancer cell lines [11] and xenograft models [8], this has failed to translate to clinical scenarios. A phase III trial showed no difference in PFS or OS with the addition of sorafenib to gemcitabine alone [25]. This was also demonstrated in a phase II trial comparing the addition of sorafenib to cisplatin/gemcitabine versus cisplatin/gemcitabine alone which showed no increase in PFS or OS [26]. Although our study shows the combination of 2DOC and sorafenib produces good response rates and durability of response, the observed rates were comparable to other FOLFOX-like regimens given in pancreatic cancer [18]. Therefore, it remains unclear how much sorafenib added to the efficacy of 2DOC.
Given the dismal outcomes associated with advanced pancreatic cancer, development of safe, efficacious, and convenient regimens remains imperative. Given the proven efficacy for first-line regimens like gemcitabine and nab-paclitaxel or FOLFIRINOX, more studies of second-line treatments are needed. Novel agents such as the oral tyrosine kinase inhibitors have shown promise in gastrointestinal cancers. However, their toxicity profile can make them difficult to combine with other chemotherapeutic agents due to overlapping toxicities. In this study, we utilized the lack of hand-foot syndrome seen in 2DOC, a derivative of modified FOLFOX6, to successfully combine it with the oral multikinase inhibitor sorafenib. This combination was safe and effective. This study adds further support to an oxaliplatin-based regimen after progression on a gemcitabine regimen for metastatic or advanced pancreatic adenocarcinoma. However, sorafenib did not appear to improve the efficacy of capecitabine and oxaliplatin for pancreas cancer as compared to RR and OS observed in other studies utilizing FOLFOX-like regimens. Possible avenues for future study include utilizing 2DOC for novel agents that have hand-foot syndrome as a common side effect but which may have activity in pancreas cancer. The optimal fluoropyrimidine regimen for gemcitabine refractory pancreas cancer should be explored in future cooperative group and phase III studies.
References
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin. doi:10.3322/caac.21254
Von Hoff DD, Ervin T, Arena FP et al (2013) Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369:1691–1703. doi:10.1056/NEJMoa1304369
Conroy T, Desseigne F, Ychou M et al (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364:1817–1825. doi:10.1056/NEJMoa1011923
Cheeseman SL, Joel SP, Chester JD et al (2002) A “modified de Gramont” regimen of fluorouracil, alone and with oxaliplatin, for advanced colorectal cancer. Br J Cancer 87:393–399. doi:10.1038/sj.bjc.6600467
Scheithauer W, Kornek GV, Raderer M et al (2002) Intermittent weekly high-dose capecitabine in combination with oxaliplatin: a phase I/II study in first-line treatment of patients with advanced colorectal cancer. Ann Oncol 13:1583–1589
Van Cutsem E, Hoff PM, Harper P et al (2004) Oral capecitabine vs intravenous 5-fluorouracil and leucovorin: integrated efficacy data and novel analyses from two large, randomised, phase III trials. Br J Cancer 90:1190–1197. doi:10.1038/sj.bjc.6601676
Mulkerin D, LoConte NK, Holen KD et al (2009) A phase I study of an oral simulated FOLFOX with high dose capecitabine. Invest New Drugs 27:461–468. doi:10.1007/s10637-008-9210-8
Wilhelm SM, Adnane L, Newell P et al (2008) Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther 7:3129–3140. doi:10.1158/1535-7163.MCT-08-0013
Hruban RH, van Mansfeld AD, Offerhaus GJ et al (1993) K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol 143:545–554
Wilhelm SM, Carter C, Tang L et al (2004) BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64:7099–7109. doi:10.1158/0008-5472.CAN-04-1443
Ulivi P, Arienti C, Amadori D et al (2009) Role of RAF/MEK/ERK pathway, p-STAT-3 and Mcl-1 in sorafenib activity in human pancreatic cancer cell lines. J Cell Physiol 220:214–221. doi:10.1002/jcp.21753
Escudier B, Eisen T, Stadler WM et al (2007) Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 356:125–134. doi:10.1056/NEJMoa060655
Lubner SJ, Loconte NK, Holen KD et al (2010) A phase II study of oxaliplatin, 5-fluorouracil, leucovorin, and high-dose capecitabine in patients with metastatic colorectal cancer. Clin Colorectal Cancer 9:157–161. doi:10.3816/CCC.2010.n.021
LoConte NK, Holen KD, Schelman WR et al (2013) A phase I study of sorafenib, oxaliplatin and 2 days of high dose capecitabine in advanced pancreatic and biliary tract cancer: a Wisconsin oncology network study. Invest New Drugs 31:943–948. doi:10.1007/s10637-012-9916-5
Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247. doi:10.1016/j.ejca.2008.10.026
CTEP (2006) Common Terminology Criteria for Adverse Events v3.0 (CTCAE). http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed 9 June 2015
Rahma OE, Duffy A, Liewehr DJ et al (2013) Second-line treatment in advanced pancreatic cancer: a comprehensive analysis of published clinical trials. Ann Oncol 24:1972–1979. doi:10.1093/annonc/mdt166
Berk V, Ozdemir N, Ozkan M et al (2012) XELOX vs. FOLFOX4 as second line chemotherapy in advanced pancreatic cancer. Hepatogastroenterology 59:2635–2639. doi:10.5754/hge12181
Cartwright TH (2014) Use of first-line chemotherapy for advanced pancreatic cancer: FOLFIRINOX versus gemcitabine-based therapy. J Clin Oncol 32:5s (suppl; abstr 4132)
Zaanan A, Trouilloud I, Markoutsaki T et al (2014) FOLFOX as second-line chemotherapy in patients with pretreated metastatic pancreatic cancer from the FIRGEM study. BMC Cancer 14:441. doi:10.1186/1471-2407-14-441
Yoo C, Hwang JY, Kim J-E et al (2009) A randomised phase II study of modified FOLFIRI.3 vs modified FOLFOX as second-line therapy in patients with gemcitabine-refractory advanced pancreatic cancer. Br J Cancer 101:1658–1663. doi:10.1038/sj.bjc.6605374
Baselga J, Segalla JGM, Roché H et al (2012) Sorafenib in combination with capecitabine: an oral regimen for patients with HER2-negative locally advanced or metastatic breast cancer. J Clin Oncol 30:1484–1491. doi:10.1200/JCO.2011.36.7771
Tabernero J, Garcia-Carbonero R, Cassidy J et al (2013) Sorafenib in combination with oxaliplatin, leucovorin, and fluorouracil (modified FOLFOX6) as first-line treatment of metastatic colorectal cancer: the RESPECT trial. Clin Cancer Res 19:2541–2550. doi:10.1158/1078-0432.CCR-13-0107
Lévy E, Piedbois P, Buyse M et al (1998) Toxicity of fluorouracil in patients with advanced colorectal cancer: effect of administration schedule and prognostic factors. J Clin Oncol 16:3537–3541
Gonçalves A, Gilabert M, François E et al (2012) BAYPAN study: a double-blind phase III randomized trial comparing gemcitabine plus sorafenib and gemcitabine plus placebo in patients with advanced pancreatic cancer. Ann Oncol 23:2799–2805. doi:10.1093/annonc/mds135
Cascinu S, Berardi R, Sobrero A et al (2014) Sorafenib does not improve efficacy of chemotherapy in advanced pancreatic cancer: a GISCAD randomized phase II study. Dig Liver Dis 46:182–186. doi:10.1016/j.dld.2013.09.020
Acknowledgments
This study was funded by Sanofi-aventis, Bayer/Onyx pharmaceuticals, and University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Makielski, R.J., Lubner, S.J., Mulkerin, D.L. et al. A phase II study of sorafenib, oxaliplatin, and 2 days of high-dose capecitabine in advanced pancreas cancer. Cancer Chemother Pharmacol 76, 317–323 (2015). https://doi.org/10.1007/s00280-015-2783-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-015-2783-y