
Abstract
Background
Reduced CYP2D6 metabolism and low Z-endoxifen (ENDX) concentrations may increase the risk of breast cancer recurrence in tamoxifen (TAM)-treated women. Little is known regarding the differences between TAM and ENDX murine pharmacokinetics or the effect of administration route on plasma concentrations of each drug.
Methods
The pharmacokinetics of TAM and ENDX were characterized in female mice.
Results
For subcutaneous [s.c.] and oral TAM (4, 10 and 20 mg/kg), TAM AUC increased in a linear manner, but concentrations of the active metabolites [ENDX and 4-hydroxytamoxifen (4HT)] remained low. For oral TAM (20 mg), 4HT concentrations were tenfold greater (>25 ng/ml) than achievable in TAM-treated humans. Both oral (10–200 mg/kg) and s.c. (2.5–25 mg/kg) ENDX·HCl resulted in a greater than dose-proportional increase in AUC, with eightfold greater ENDX concentrations than an equivalent TAM dose. ENDX accumulated in plasma after 5-day dosing of 25 or 100 mg/kg ENDX·HCl and exceeded target concentrations of 0.1 and 1.0 μM, respectively, by twofold to fourfold.
Conclusions
In murine models, oral ENDX yields substantially higher ENDX concentrations, compared to TAM. The low 4HT and ENDX concentrations observed in mice receiving s.c. TAM mirror the TAM pharmacokinetics in humans with impaired CYP2D6 metabolism. These data support the ongoing development of ENDX as a novel agent for the endocrine treatment of ER-positive breast cancer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
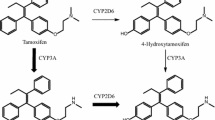
Tamoxifen (TAM) is a weak anti-estrogen that undergoes extensive biotransformation in humans to metabolites that have far greater anti-estrogenic potency. 4-Hydroxytamoxifen (4HT) represents less than 10 % of TAM primary oxidation [1, 2], but has 100-fold greater affinity for the estrogen receptor (ER) and 30-fold to 100-fold greater potency in suppressing estrogen-dependent cell proliferation compared with TAM [3–5]. The secondary TAM metabolite Z-N-desmethyl-4-hydroxytamoxifen (ENDX) has equivalent in vitro ER-binding potency [6] and suppression of ER-dependent human breast cancer cell proliferation compared to 4HT [6, 7], and is thought to substantially contribute to TAM activity [8].
In humans, ENDX plasma concentrations are typically sixfold–tenfold higher [9–11] than 4HT plasma concentrations (<10 nM), but exhibit substantial variability that is partially explained by genetic and drug-induced variation in CYP2D6 enzyme activity. TAM-treated women with reduced or absent CYP2D6 activity have low concentrations of ENDX [10–13]. While controversy continues to exist regarding the role of CYP2D6 and TAM in clinical outcomes [14, 15], recent secondary analyses from independent large prospective studies [16, 17], as well as data from the metastatic setting [18], have demonstrated that both CYP2D6 genotype [17] and ENDX concentrations [16, 17] are associated with disease-free survival. Based on these observations, studies are ongoing to develop endoxifen in patients with hormone refractory cancer (NCT01327781 and NCT01273168).
Because primary and secondary metabolism limits the concentrations of the active metabolites of TAM, we hypothesized that clinically relevant concentrations and exposures of ENDX could be achieved following direct oral administration of ENDX. Therefore, we characterized and compared the pharmacokinetics of intravenous (i.v.) and oral ENDX with oral and subcutaneous (s.c.) TAM in female mice.
Materials and methods
Z-Endoxifen hydrochloride (ENDX·HCl) was provided by the Developmental Therapeutics Program, NCI. TAM, 4HT, N-desmethyltamoxifen (NDMT), and toremifene (a selective ER modulator used as the internal standard) were purchased from Sigma Chemical Company (St. Louis, MO). The primary stock solutions of ENDX (2 mg/ml), 4HT (1 mg/ml), NDMT (1 mg/ml), TAM (1 mg/ml), and toremifene (1 mg/ml) were prepared in ethanol and stored at −20 °C. Working standard solutions were prepared in ethanol and stored at −20 °C. All animal studies were performed under protocols and guidelines approved by the Institutional Animal Care and Use Committee of the Mayo Clinic (Rochester, MN). Female CD1 mice were supplied by Charles River (Portage, MI). Female ovariectomized BALB/c athymic nude mice were obtained from Harlan Laboratories (Indianapolis, IN). The mice were housed in the Department of Comparative Medicine Animal Facility, which provided a pathogen-free environment under controlled conditions of light and humidity, and accredited by the International Association for Assessment and Accreditation of Laboratory Animal Care. Mice were acclimatized for 48 h after arrival and received food and water ad libitum.
HPLC assay
Plasma concentrations of TAM, ENDX, 4HT, NDMT were measured using a modification of a previously published HPLC assay [19] that utilized a concave gradient elution profile and a post-column photochemical reactor (PHRED-8, Aura Industries) with fluorescence detection (excitation wavelength—250 nm, emission wavelength—370 nm). This assay has been validated in our laboratory according to FDA guidance. The lower limit of quantification and linear range were 1 ng/ml and 1–500 ng/ml, respectively. In brief, TAM, ENDX, 4HT, NDMT, and toremifene were isolated from plasma samples using liquid–liquid extraction. Dried extracts were reconstituted in 100 μl of 55:45 ACN:20 mM KH2PO4, pH 3.0, transferred to an amber auto-sampler vial with glass insert and placed in an autosampler maintained at 4 °C. Compounds were separated on a Genesis C18 (250 mm × 3.0 mm, 3 µ particle size) analytical column protected by a Genesis C18 (10 mm × 3.0 mm, 3 µ particle size) pre-column using a concave gradient elution profile from 43:57 (v/v) ACN:20 mM KH2PO4, pH 3.0, to 75:25 (v/v) ACN:20 mM KH2PO4, pH 3.0 (flow rate—0.5 ml/min, injection volume—50 μl). All procedures were performed in the dark under minimum exposure to light.
Mouse pharmacokinetic studies
Pharmacokinetics were characterized in female CD1 and ovariectomized BALB/c athymic nude mice. Blood samples were collected from mice anesthetized under isoflurane by cardiac puncture into a syringe containing 0.15 ml of 10 % heparin/CPDA anticoagulant and transferred to silanized amber microcentrifuge tubes. Plasma and red blood cells were separated immediately by centrifugation in a refrigerated centrifuge set at 4 °C. Plasma was transferred to a separate tube, and both tubes were immediately frozen at −20 °C until analysis.
Single-dose pharmacokinetics in CD1 mice
For i.v. dosing, ENDX·HCl was dissolved in 5 % ethanol, 5 % PEG 400, and 90 % normal saline (final concentration—0.2 mg/ml ENDX) for the 1 mg/kg i.v. dose. Blood samples (2–6 mice per time-point) were collected 5, 15 min, 0.5, 1, 2, 4, 8, 16, and 24 h after administering the dose. For oral dosing, TAM (4, 10, and 20 mg/kg) and ENDX·HCl (10, 25, 50, 75, and 200 mg/kg) were suspended in 0.5 % carboxymethyl cellulose (CMC). Blood samples (three mice per time-point) were collected 0.5, 1, 2, 4, 8, 16, and 24 h after administering the dose. For subcutaneous dosing, TAM (4, 10, and 20 mg/kg) was suspended in 0.5 % CMC and ENDX·HCl (2.5 and 25 mg/kg) was prepared in 1 mM ascorbic acid:PEG400, 1:1 v/v. Blood samples (three mice per time-point) were collected 0.5, 1, 2, 4, 8, 16, and 24 h after administering the dose.
Multiple-dose pharmacokinetics in CD1 mice
ENDX·HCl (25 or 100 mg/kg) was suspended in 0.5 % CMC and administered orally for five consecutive days. Blood samples (five mice per time-point) were collected 4 h after administering the dose on day 1 and day 5.
Single-dose pharmacokinetics in ovariectomized female nude mice
Ovariectomized female BALB/c athymic nude mice 4–6 weeks of age supplemented with 1.7-mg 90-day-release estrogen pellets (17β-estradiol, NE-121, Innovative Research, Novi, MI). Mice were randomly assigned to receive oral TAM (75 mg, suspended in 0.3 % hydroxypropyl cellulose) or oral ENDX (75 mg/kg dissolved in 3 mM PEG400:ascorbic acid, 50:50 v/v). A third group of female CD1 mice received oral ENDX (75 mg/kg dissolved in PEG400:ascorbic acid, 50:50 v/v). Blood samples (three mice per time-point) were collected before treatment and 1, 4, 8, and 24 h after administering the dose.
Data analysis
Pharmacokinetics were estimated by standard non-compartmental analysis methods using WinNonlin (Professional Version 3.0; Pharsight Corp; Mountain View, CA). The apparent terminal elimination rate constants (λz) were determined by linear least-squares regression through the linear terminal portion of the graph of the log plasma concentration versus time. The apparent elimination half-life (t 1/2) was calculated as 0.693/λz. Area under the plasma concentration–time curve (AUC0–T ) was determined using the linear trapezoidal rule from time zero to the time of the last detectable sample (T). Area under the plasma concentration–time curves through infinite time (AUC0–∞) was calculated by adding C T /λz to AUC0–T . The CLp was calculated as dose/AUC0–∞. Bioavailability was calculated as follows: (AUCpo/AUCiv)·(Doseiv/Dosepo)·100 %.
Differences in concentration levels between groups were assessed using Kruskal–Wallis test. A p value <0.05 was considered significant.
Results
The pharmacokinetics of TAM and its major circulating metabolites were characterized in female CD1 mice administered s.c. and oral doses of 4, 10, and 20 mg/kg, which are the most commonly utilized doses for in vivo studies [20]. The pharmacokinetic data are summarized in Table 1. Plasma profiles for oral and s.c. administration of 20 mg/kg TAM are shown in Fig. 1a, b, respectively.

Plasma profiles for average (n = 3) TAM (closed circle), 4HT (open circle), NDMT (closed inverted triangle) and ENDX (open triangle) following a s.c. and b oral administration of 500 μg (20 mg/kg) TAM
For both the oral and s.c. TAM doses, plasma concentrations increased significantly across dose levels (oral: p values ≤0.044 for 0.5, 1, 2, 4, and 16 h; s.c.: p values ≤0.039 for 0.5, 2, 4, 16, and 24 h). At the 20 mg/kg dose, TAM peak plasma concentrations (C max) were achieved 0.5–1 h after oral administration; however, only one of the seven mice achieved C max > 30.0 ng/ml. TAM C max values were achieved 0.5–4 h after s.c. administration, and 11 of 12 mice achieved C max > 30.0 ng/ml. TAM AUC increased with dose; however, comparing equivalent oral and s.c. doses, TAM AUC values following oral administration were 2.5–14 % of the AUC values achieved following s.c. administration (Table 1).
Detection of active TAM metabolites in plasma was dependent on the TAM dose and route of administration. Metabolite concentrations of 4HT and ENDX were consistently higher following oral administration compared to s.c. administration. 4HT was detected after each dose and both routes, but concentrations were low (C max, 2.0–7.3 ng/ml) after s.c. dosing. ENDX was not detected until the 20 mg/kg s.c. dose and 10 and 20 mg/kg oral doses. At the 20 mg/kg dose, plasma concentrations of 4HT and ENDX were lower for the s.c. compared to the oral route of administration, and at 1 h, this difference was statistically significant (p = 0.0339 and p = 0.0323, respectively).
Since low ENDX concentrations of were produced following s.c. and oral TAM, we investigated the pharmacokinetics and bioavailability of ENDX after direct dosing of this active TAM metabolite (Table 2). Following an i.v. dose of 1 mg/kg ENDX·HCl, a graph of the plasma ENDX concentration versus time showed rapid distribution and slower elimination (Fig. 2). The ENDX C max, terminal half-life and plasma clearance values were 96 ng/ml, 6.5 h, and 11.8 l/h/kg, respectively.

ENDX plasma profile after a single i.v. dose of 1 mg/kg ENDX·HCl
After oral administration, ENDX C max was achieved within 0.25–2 h. Plasma concentrations increased significantly across dose levels (10, 25, 50, 75, and 200 mg/kg) at 0.5, 1, 2, and 4 h after oral administration (p value ≤0.001). As the dose was increased from 10 to 200 mg/kg, the C max and AUC values increased 88-fold and 400-fold, respectively. Apparent oral bioavailability increased from 12 to >200 % over this same dose range. Moreover, plasma concentrations exceeding 1 μM could be achieved and maintained for 24 h following a single oral dose of 75 mg/kg ENDX·HCl. Thus, high plasma concentrations, associated with previously observed in vitro ENDX cytotoxicity [8], were achieved and maintained after a single oral ENDX·HCl dose.
The pharmacokinetic data for ENDX following equivalent s.c. and oral doses of 20 mg/kg TAM and 25 mg/kg ENDX·HCl are shown in Tables 1 and 2, respectively. ENDX C max and AUC following oral ENDX·HCl (103 ng/ml and 660 ng/ml h, respectively; Table 2) were eightfold higher than those same values following the equivalent oral dose of TAM (13.4 ng/ml and 83.1 ng/ml h; Table 1). ENDX C max and AUC following s.c. ENDX·HCl (935 ng/ml and 4,920 ng/ml h, respectively; Table 2) were >140-fold higher than those same values following the equivalent oral dose of TAM (1.9 ng/ml and 34.6 ng/ml h; Table 1). Finally, when comparing oral with s.c. ENDX·HCl dosing, C max and AUC values achieved with a 2.5 mg/kg s.c. dose were similar to those achieved with 10–25 mg/kg oral doses (Table 2).
Based on the single-dose pharmacokinetic data, we selected ENDX·HCl doses of 25 and 100 mg/kg for multiple oral dose regimens to yield ENDX plasma concentrations of 100 nM (37 ng/ml) and 1–2 μM (370–740 ng/ml), respectively. The ENDX concentrations were measured 4 h after the daily dose on day 1 and day 5. As shown in Table 3, ENDX accumulated in plasma after 5-day dosing and this difference was significant at the 100 mg/kg dose (Wilcoxon rank sum test p = 0.036).
To confirm the similarity of ENDX and TAM pharmacokinetics between CD1 mice and nude mice used for tumor xenograft studies, we determined ENDX C max and AUC0–24h values in CD1 mice and tumor-bearing nude mice treated with an oral dose of 75 mg/kg ENDX·HCl or TAM. The ENDX C max and AUC0–24h values for tumor-bearing nude mice (1,030 ng/ml and 15,500 ng/ml h) were similar to the C max and AUC0–24h values for CD1 mice (888 ng/ml and 12,700 ng/l h) and were 27-fold higher than the ENDX C max and AUC0–24h values (33 ng/ml and 471 ng/ml h) following TAM administration.
Discussion
Tamoxifen is an effective and widely used drug for treatment of ER+ breast cancer. While TAM itself has both estrogenic and antiestrogenic activity, it is a prodrug with complex metabolism [21]. Cytochrome P450-catalyzed oxidation yields several hydroxylated metabolites including 4HT and ENDX, which have much greater anti-estrogenic activity than the parent drug [8, 21]. In TAM-treated humans, plasma concentrations of 4HT are low (<5 ng/ml) [21]. In marked contrast, plasma concentrations of ENDX are up to tenfold higher than 4HT, but vary widely (3–26 ng/ml) based in part on CYP2D6 polymorphisms and concomitant administration of drugs that inhibit CYP2D6 enzyme activity [21].
Given the importance of ENDX as it relates to disease-free survival in human TAM-treated breast cancer [16], recent data on the pharmacologic activity of TAM metabolites [8, 21], as well as differences in P450 enzyme isoform expression in mice and humans [22, 23], it is essential to reexamine the murine pharmacokinetics of tamoxifen. This point is critical since the current standard is to use 4HT to model the in vitro antitumor activity of TAM, but to use s.c. administration of the parent drug, TAM for in vivo studies. This is based in part on a head-to-head comparison of s.c. TAM with s.c. mono-hydroxytamoxifen where TAM was more effective than mono-hydroxytamoxifen in an in vivo DMBA-induced rat mammary carcinoma model [24].
Therefore, our first goal was to characterize the murine pharmacokinetics of the major TAM metabolites following oral and s.c. administration. An early study of TAM pharmacokinetics in mice using high oral doses (50 and 200 mg/kg) demonstrated dose-dependent formation of NDMT and 4HT, exposure to higRh concentrations of 4HT that did not reflect pharmacokinetics in humans, and trace concentrations of ENDX that could not be quantified due to the lack of pure compound [25]. Here, we demonstrate that the pharmacokinetics of TAM and 4HT are consistent with earlier studies, with very low 4HT concentrations detected after s.c. dosing and much higher 4HT concentrations observed after oral dosing. In fact, s.c. administration of 20 mg/kg TAM to female mice yields low exposure to 4HT (7.3 ng/ml) and ENDX (1.9 ng/ml), and mirrors the concentrations of active metabolites observed in TAM-treated humans with deficient CYP2D6 metabolism. The 20 mg/kg oral dose yielded a tenfold higher 4HT (26.4 ng/ml) than observed in humans receiving an oral TAM dose of 20 mg/day. We observed much lower NDMT concentrations than either TAM or 4HT. The reason for the low NDMT is unclear, but may be due to differences in dose or variability in cytochrome P4503A enzyme expression across mouse strains including CD1 mice [23]. Importantly, with the availability of a pure standard for ENDX, we were able to confirm the presence of very low ENDX levels.
Our second goal was to select the route and dose of TAM that most closely resembled the concentrations of the active metabolites in TAM-treated humans, and to compare concentrations of 4HT and ENDX based upon the route of TAM administration (oral vs. s.c.). Our observation that 4HT and ENDX concentrations were substantially lower following s.c. compared with oral administration is consistent with the fact that the s.c. route circumvents first-pass metabolism and results in lower systemic availability of the active metabolites. Specifically, peak concentrations of 4HT and ENDX were 7.3 and 1.9 ng/ml, respectively, following a single s.c. dose of 20 mg/kg TAM, and very similar to the concentrations observed in humans with deficient CYP2D6 metabolism. In contrast, oral administration of TAM resulted in a 4HT concentration that was twice that of ENDX (26.4 and 13.4 ng/ml, respectively) and tenfold higher than known to be achievable in TAM-treated humans. This difference is likely due to the lower expression of CYP2D and CYP3A enzymes in mice compared to humans [22] and differences in P450-catalyzed metabolism of TAM between humans and mice. CYP2D6 was originally characterized as debrisoquine hydroxylase in rat and human liver, but does not appear to be homologous to the P450 enzyme that catalyzes debrisoquine hydroxylation in mice since it is not inhibited by an antibody against purified rat liver CYP2D that inhibits debrisoquine hydroxylase activity in rat liver microsomes [23, 26]. Consequently, mouse debrisoquine hydroxylase either is a mouse CYP2D enzyme that is not recognized by anti-rat CYP2D or belongs to another P450 subfamily. Therefore, oral TAM disposition in mice is not representative of human oral TAM disposition, and this route should not be used to model human TAM antitumor activity.
To overcome the limitations of primary and secondary metabolism necessary for the metabolic activation of TAM in humans, we investigated the ENDX pharmacokinetics. A single i.v. dose of 1 mg/kg ENDX·HCl achieved a peak concentration that was more than twofold higher than the concentration associated with activity against breast cancer in women treated with TAM [16]. A single oral dose of 10 mg/kg ENDX·HCl achieved a peak concentration associated with maximal in vitro activity [8]. Clinically active concentrations of ENDX were sustained for 8 h with a single dose of 50 mg/kg ENDX·HCl, and concentrations greater than 1 μM (370 ng/ml) were sustained for 24 h with a single dose of 75 mg/kg ENDX·HCl.
Two prior studies have evaluated the in vivo antitumor activity of ENDX [27, 28]. In the first study, Ahmed et al. evaluated low doses of oral ENDX (2–8 mg) compared with oral TAM (20 mg) using MCF7 xenografts and observed that both TAM and ENDX suppressed antitumor activity, but demonstrated no difference comparing the 8 mg oral ENDX dose with the 20 mg oral TAM dose. Pharmacokinetics were not evaluated in either the TAM or ENDX treated mice, and given our observations that oral TAM results in high concentrations of 4HT, the antitumor activity in oral TAM-treated mice is likely due to 4HT with a small contribution from ENDX. Recently, Gong et al. evaluated the relationship between ENDX exposure and tumor growth inhibition and demonstrated 100 % growth inhibition in MCF7 xenografts using a 30–60 mg/kg dose of ENDX. While Gong et al. did not compare ENDX with TAM, it should be noted that the doses, concentrations, and antitumor activity of ENDX were similar to our findings and suggest that the optimal concentrations of ENDX needed to inhibit antitumor activity appear to be higher than those achievable in TAM-treated humans.
Of note, the data from our studies with mice show evidence of dose-dependent pharmacokinetics as illustrated by greater than dose-proportional increases in AUC and C max over the dose range studied (Table 2). There are two possible explanations for dose-dependent pharmacokinetics of ENDX. First, TAM and its primary metabolites, NDMT and 4HT, are effective inhibitors of CYP3A activity [29]. The low K i values for reversible CYP3A inhibition by NDMT and 4HT, as well as the low K i values for mechanism-based CYP3A inhibition by TAM and NDMT, are similar to the steady-state plasma of those found when TAM is administered at doses of 20–40 mg/day. By analogy, it can be hypothesized that CYP3A activity may be inhibited by ENDX at concentrations achievable following oral doses. Thus, it is possible that inhibition of CYP3A in small intestine epithelium would increase drug absorption, while inhibition of CYP3A activity in the liver would decrease hepatic clearance. Second, higher doses may inhibit drug transporters involved in ENDX absorption and clearance. Several investigators have examined the role of p-glycoprotein in the disposition of TAM. TAM, NDMT, and 4HT were inhibitors, but not substrates, for p-glycoprotein [30]. 4HT was the most potent inhibitor with an IC50 value that was threefold lower than the values observed for TAM and NDMT. In an in vivo study quercetin, an inhibitor of p-glycoprotein and CYP3A activity increased the rate of absorption and AUC of TAM, increased the 4HT AUC, and decreased the 4HT/TAM ratio [31]. These observations were consistent with an effect of quercetin on both CYP3A and p-glycoprotein. Similarly, ENDX has been shown to be a substrate for p-glycoprotein in both in vitro studies of p-glycoprotein-dependent flux across LLCPK cells that overexpressed p-glycoprotein and in vivo studies of plasma and brain disposition of ENDX in Mdr1a-deficient mice [32]. The result of both mechanisms would be to increase systematic exposure and apparent bioavailability of ENDX.
Conclusions
In conclusion, our studies provide important insight into the murine pharmacokinetics of both TAM and ENDX, and demonstrate the importance of dose and route of administration as it relates to the concentrations of the active metabolites. These data further suggest that the administration of ENDX may result in greater antitumor activity by means of increased ENDX exposure and provide support for the ongoing development of ENDX as a new hormonal therapy for ER+ breast cancer. Based on these data and in collaboration with the National Cancer Institute, phase I and II clinical trials of ENDX are ongoing/planned in women with ER-positive breast cancer and in patients with hormonally positive tumors (NCT01327781 and NCT01273168).
Abbreviations
- CYP2D6:
-
Cytochrome P450
- ENDX:
-
Endoxifen (4-hydroxy-N-desmethyl tamoxifen)
- TAM:
-
Tamoxifen
- 4HT:
-
4-Hydroxytamoxifen
- NDMT:
-
N-Desmethyltamoxifen
- ER:
-
Estrogen receptor
- s.c.:
-
Subcutaneous
- i.v.:
-
Intravenous
- HPLC:
-
High-pressure liquid chromatography
- ACN:
-
Acetonitrile
- KH2PO4 :
-
Monopotassium phosphate
- PEG:
-
Polyethylene glycol
- CMC:
-
Carboxymethylcellulose
- CPDA:
-
Citrate/phosphate/dextrose/adenine anticoagulant solution
- AUC:
-
Area under the plasma concentration–time curve
- C max :
-
Maximum plasma concentration
- T max :
-
Time that the maximum plasma concentration was achieved
- Vz:
-
Volume of distribution
- Clp:
-
Plasma clearance
References
Lonning PE, Lien EA, Lundgren S, Kvinnsland S (1992) Clinical pharmacokinetics of endocrine agents used in advanced breast cancer. Clin Pharmacokinet 22:327–358
Desta Z, Ward BA, Soukhova NV, Flockhart DA (2004) Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6. J Pharmacol Exp Ther 310:1062–1075
Borgna JL, Rochefort H (1981) Hydroxylated metabolites of tamoxifen are formed in vivo and bound to estrogen receptor in target tissues. J Biol Chem 256:859–868
Robertson DW, Katzenellenbogen JA, Long DJ, Rorke EA, Katzenellenbogen BS (1982) Tamoxifen antiestrogens. A comparison of the activity, pharmacokinetics, and metabolic activation of the cis and trans isomers of tamoxifen. J Steroid Biochem 16:1–13
Jordan VC (1982) Metabolites of tamoxifen in animals and man: identification, pharmacology, and significance. Breast Cancer Res Treat 2:123–138
Johnson MD, Zuo H, Lee KH, Trebley JP, Rae JM, Weatherman RV, Desta Z, Flockhart DA, Skaar TC (2004) Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Res Treat 85:151–159
Lim YC, Desta Z, Flockhart DA, Skaar TC (2005) Endoxifen (4-hydroxy-N-desmethyl-tamoxifen) has anti-estrogenic effects in breast cancer cells with potency similar to 4-hydroxy-tamoxifen. Cancer Chemother Pharmacol 55:471–478
Wu X, Hawse JR, Subramaniam M, Goetz MP, Ingle JN, Spelsberg TC (2009) The tamoxifen metabolite, endoxifen, is a potent antiestrogen that targets estrogen receptor alpha for degradation in breast cancer cells. Cancer Res 69:1722–1727
Stearns V, Johnson MD, Rae JM, Morocho A, Novielli A, Bhargava P, Hayes DF, Desta Z, Flockhart DA (2003) Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst 95:1758–1764
Jin Y, Desta Z, Stearns V, Ward B, Ho H, Lee KH, Skaar T, Storniolo AM, Li L, Araba A, Blanchard R, Nguyen A, Ullmer L, Hayden J, Lemler S, Weinshilboum RM, Rae JM, Hayes DF, Flockhart DA (2005) CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst 97:30–39
Borges S, Desta Z, Li L, Skaar TC, Ward BA, Nguyen A, Jin Y, Storniolo AM, Nikoloff DM, Wu L, Hillman G, Hayes DF, Stearns V, Flockhart DA (2006) Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin Pharmacol Ther 80:61–74
Gjerde J, Hauglid M, Breilid H, Lundgren S, Varhaug JE, Kisanga ER, Mellgren G, Steen VM, Lien EA (2008) Effects of CYP2D6 and SULT1A1 genotypes including SULT1A1 gene copy number on tamoxifen metabolism. Ann Oncol 19:56–61
Stearns V, Beebe KL, Iyengar M, Dube E (2003) Paroxetine controlled release in the treatment of menopausal hot flashes: a randomized controlled trial. JAMA 289:2827–2834
Rae JM, Drury S, Hayes DF, Stearns V, Thibert JN, Haynes BP, Salter J, Sestak I, Cuzick J, Dowsett M (2012) CYP2D6 and UGT2B7 genotype and risk of recurrence in tamoxifen-treated breast cancer patients. J Natl Cancer Inst 104:452–460
Regan MM, Leyland-Jones B, Bouzyk M, Pagani O, Tang W, Kammler R, Dell’orto P, Biasi MO, Thurlimann B, Lyng MB, Ditzel HJ, Neven P, Debled M, Maibach R, Price KN, Gelber RD, Coates AS, Goldhirsch A, Rae JM, Viale G (2012) CYP2D6 genotype and tamoxifen response in postmenopausal women with endocrine-responsive breast cancer: the breast international group 1-98 trial. J Natl Cancer Inst 104:441–451
Madlensky L, Natarajan L, Tchu S, Pu M, Mortimer J, Flatt SW, Nikoloff DM, Hillman G, Fontecha MR, Lawrence HJ, Parker BA, Wu AH, Pierce JP (2011) Tamoxifen metabolite concentrations, CYP2D6 genotype, and breast cancer outcomes. Clin Pharmacol Ther 89:718–725
Goetz MP, Suman VJ, Hoskin TL, Gnant M, Filipits M, Safgren SL, Kuffel M, Jakesz R, Rudas M, Greil R, Dietze O, Lang A, Offner F, Reynolds CA, Weinshilboum RM, Ames MM, Ingle JN (2013) CYP2D6 metabolism and patient outcome in the austrian breast and colorectal cancer study group trial (abcsg) 8. Clin Cancer Res 19:500–507
Karle J, Bolbrinker J, Vogl S, Kreutz R, Denkert C, Eucker J, Wischnewsky M, Possinger K, Regierer AC (2013) Influence of CYP2D6-genotype on tamoxifen efficacy in advanced breast cancer. Breast Cancer Res Treat 139:553–560
Lee KH, Ward BA, Desta Z, Flockhart DA, Jones DR (2003) Quantification of tamoxifen and three metabolites in plasma by high-performance liquid chromatography with fluorescence detection: application to a clinical trial. J Chromatogr 791:245–253
Schiff R, Reddy P, Ahotupa M, Coronado-Heinsohn E, Grim M, Hilsenbeck SG, Lawrence R, Deneke S, Herrera R, Chamness GC, Fuqua SA, Brown PH, Osborne CK (2000) Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo. J Natl Cancer Inst 92:1926–1934
Murdter TE, Schroth W, Bacchus-Gerybadze L, Winter S, Heinkele G, Simon W, Fasching PA, Fehm T, Eichelbaum M, Schwab M, Brauch H (2011) Activity levels of tamoxifen metabolites at the estrogen receptor and the impact of genetic polymorphisms of phase I and II enzymes on their concentration levels in plasma. Clin Pharmacol Ther 89:708–717
McLaughlin LA, Dickmann LJ, Wolf CR, Henderson CJ (2008) Functional expression and comparative characterization of nine murine cytochromes P450 by fluorescent inhibition screening. Drug Metab Dispos 36:1322–1331
Lofgren S, Hagbjork AL, Ekman S, Fransson-Steen R, Terelius Y (2004) Metabolism of human cytochrome P450 marker substrates in mouse: a strain and gender comparison. Xenobiotica 34:811–834
Jordan VC, Allen KE (1980) Evaluation of the antitumour activity of the non-steroidal antioestrogen monohydroxytamoxifen in the DMBA-induced rat mammary carcinoma model. Eur J Cancer 16:239–251
Robinson SP, Langan-Fahey SM, Jordan VC (1989) Implications of tamoxifen metabolism in the athymic mouse for the study of antitumor effects upon human breast cancer xenografts. Eur J Cancer Clin Oncol 25:1769–1776
Masubuchi Y, Iwasa T, Hosokawa S, Suzuki T, Horie T, Imaoka S, Funae Y, Narimatsu S (1997) Selective deficiency of debrisoquine 4-hydroxylase activity in mouse liver microsomes. J Pharmacol Exp Ther 282:1435–1441
Ahmad A, Ali SM, Ahmad MU, Sheikh S, Ahmad I (2010) Orally administered endoxifen is a new therapeutic agent for breast cancer. Breast Cancer Res Treat 122:579–584
Gong IY, Teft WA, Ly J, Chen YH, Alicke B, Kim RB, Choo EF (2013) Determination of clinically therapeutic endoxifen concentrations based on efficacy from human MCF7 breast cancer xenografts. Breast Cancer Res Treat 139:61–69
Zhao XJ, Jones DR, Wang YH, Grimm SW, Hall SD (2002) Reversible and irreversible inhibition of CYP3A enzymes by tamoxifen and metabolites. Xenobiotica 32:863–878
Bekaii-Saab TS, Perloff MD, Weemhoff JL, Greenblatt DJ, von Moltke LL (2004) Interactions of tamoxifen, N-desmethyltamoxifen and 4-hydroxytamoxifen with P-glycoprotein and CYP3A. Biopharm Drug Dispos 25:283–289
Shin SC, Choi JS, Li X (2006) Enhanced bioavailability of tamoxifen after oral administration of tamoxifen with quercetin in rats. Int J Pharm 313:144–149
Teft WA, Mansell SE, Kim RB (2011) Endoxifen, the active metabolite of tamoxifen, is a substrate of the efflux transporter p-glycoprotein (multidrug resistance 1). Drug Metab Dispos 39:558–562
Acknowledgments
Supported in part by 1R01CA133049-01 (MPG, MMA), the Mayo Comprehensive Cancer Center Grant (CA15083; MMA, JMR, VS), and the Mayo Clinic Breast Cancer SPORE (CA 116201; MMA, JMR, MPG, PH, XH).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Joel M. Reid and Matthew P. Goetz have contributed equally to this article.
Rights and permissions
About this article

Cite this article
Reid, J.M., Goetz, M.P., Buhrow, S.A. et al. Pharmacokinetics of endoxifen and tamoxifen in female mice: implications for comparative in vivo activity studies. Cancer Chemother Pharmacol 74, 1271–1278 (2014). https://doi.org/10.1007/s00280-014-2605-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-014-2605-7