
Abstract
Purpose
The metabolism of pazopanib is primarily mediated by CYP3A4. The solubility of pazopanib is pH-dependent, and an elevated gastric pH may decrease its bioavailability. This study evaluated the effect of a potent CYP3A4 inhibitor, ketoconazole, and the proton pump inhibitor esomeprazole on the pharmacokinetics and safety of pazopanib and its metabolites.
Methods
In Arm A, patients received pazopanib 400 mg alone once daily for 7 days followed by pazopanib 400 mg plus ketoconazole 400 mg once daily for 5 days. In Arm B, patients received pazopanib 800 mg once daily for 7 days, followed by pazopanib 800 mg plus esomeprazole 40 mg once daily for 5 days, and then pazopanib alone on the last day.
Results
Arm A enrolled 21 patients. In the presence of ketoconazole, mean area under the plasma concentration–time curve 24 h post-dose (AUC(0–24)) and mean maximum observed concentration (C max) of pazopanib increased by 66 and 45 %, respectively; mean AUC(0–24) and C max for pazopanib metabolites were lower or remained unchanged. Arm B enrolled 13 patients. In the presence of esomeprazole, mean pazopanib AUC(0–24) and C max decreased by 40 and 42 %, respectively; mean values of those parameters for metabolites of pazopanib also decreased.
Conclusions
Concomitant use of pazopanib with a strong CYP3A4 inhibitor should be avoided. If coadministration is necessary, pazopanib should be reduced to 400 mg. Concomitant use of pazopanib and proton pump inhibitors should also be avoided. Alternative dosing regimens that do not increase gastric pH at the time of pazopanib dosing should be considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pazopanib is an oral tyrosine kinase inhibitor (TKI) of vascular endothelial growth factor receptors (VEGFR-1, VEGFR-2, and VEGFR-3), platelet-derived growth factor receptors (PDGFR-α and PDGFR-β), and c-kit [1]. Pazopanib is currently approved for the treatment of patients with advanced renal cell carcinoma and patients with advanced soft tissue sarcoma who have received prior chemotherapy [2, 3]. Pazopanib 800 mg administered once daily was recommended for evaluation in phase II and III studies based on the pharmacokinetic and clinical results from the phase I trial [4]. After repeated dosing of pazopanib, the mean steady-state area under the plasma concentration–time curve 24 h post-dose (AUC(0–24)) was 743.3 μg h/mL and the median time to maximal plasma concentration (t max) was 2.0 h [4]. The mean half-life of pazopanib was 31.1 h [4]. Therefore, steady-state plasma concentrations of pazopanib are achieved after 7 days of continuous administration or by 5 half-lives.
To ensure that safe and effective plasma concentrations of pazopanib are maintained during therapy, it is important to consider the effects of other medications that could alter its absorption and metabolism. Pazopanib is metabolized primarily by the CYP450 isoform CYP3A4, with minor contributions from CYP1A2 and CYP2C8 [5]. Other TKIs predominantly metabolized by CYP3A4 include neratinib, imatinib, and dasatinib. Ketoconazole, a potent CYP3A4 inhibitor, has been shown to increase systemic exposure of these agents [6–8]. Gastric pH also may play a role in the absorption of and exposure to pazopanib. In vitro studies have revealed that in aqueous media, pazopanib is very slightly soluble at pH 1.0 and is practically insoluble above a pH of 4.0 [9]. Esomeprazole, a potent inhibitor of gastric acid secretion, is used in the treatment of gastroesophageal reflux disease and gastric and duodenal ulcers. Repeated administration of esomeprazole maintains gastric pH above 4.0 for 14 h [10]. The rate and extent of absorption of the TKI nilotinib have been shown to be reduced by esomeprazole [11].
Common adverse events (AEs) associated with pazopanib therapy include hypertension and elevated liver enzymes [2]. Severe hepatotoxicity has also been reported [12]. Concomitant administration of CYP3A4 inhibitors could lead to increased systemic exposure to pazopanib and increased risk of toxicity. Conversely, concomitant administration of drugs that raise gastric pH could potentially reduce steady-state pazopanib concentrations to subtherapeutic levels. Since many commonly used drugs are inhibitors of CYP3A4, and inhibitors of gastric acid secretion are widely prescribed, a study of drug–drug interactions with pazopanib was designed to determine the effect of repeated oral dosing of ketoconazole and esomeprazole on the pharmacokinetics of pazopanib in patients with solid tumors.
Methods
Patient eligibility
Eligible patients were at least 18 years of age with a histologic or cytologic diagnosis of advanced solid tumor that had relapsed, was refractory to standard therapy or for which there was no standard therapy. Additional eligibility criteria included Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 and adequate hematologic, hepatic, renal, and blood coagulation function. Major exclusion criteria included history or clinical evidence of central nervous system metastases, significant gastrointestinal abnormalities that could increase the risk of gastrointestinal bleeding or affect the absorption of study drugs, presence of uncontrolled infection, corrected QT interval (QTc) above 480 ms, significant cardiovascular abnormality less than 6 months before enrollment, poorly controlled hypertension, or evidence of active bleeding. Patients were prohibited from using medications that inhibit or induce CYP3A4 and drugs that increase gastric pH, to avoid confounding the study objectives. The study protocol was reviewed and approved by the institutional review board of each participating center. The trial was conducted in accordance with good clinical practice and the principles of the Declaration of Helsinki. All patients provided written informed consent before receiving treatment.
Trial design
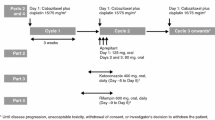
This was an open-label, two-center, two-arm trial with a two-period sequential design (VEG113971; ClinicalTrials.gov identifier NCT01205230) to assess the effects of ketoconazole (Arm A) and esomeprazole (Arm B) on the pharmacokinetics of pazopanib and its metabolites.
Arm A
During period 1, patients received pazopanib 400 mg once daily for 7 days. During period 2, patients received pazopanib 400 mg and ketoconazole 400 mg once daily for 5 days.
Arm B
During period 1, patients received pazopanib 800 mg daily in the morning for 7 days. On day 7 of period 1, patients took the first dose of esomeprazole 40 mg in the evening. During period 2, patients received pazopanib 800 mg and esomeprazole 40 mg for 4 days. On the final day of period 2, patients received only pazopanib 800 mg.
After completion of period 2 in either arm, the dose of pazopanib could be reduced or increased in increments of 200 mg, to a minimum dose of 200 mg or a maximum dose of 800 mg. A target of 12 evaluable patients was to be enrolled in each arm. Patients were considered evaluable if they received all doses during periods 1 and 2 without dose interruptions or modifications, and they had evaluable pharmacokinetic data. If no more than one patient experienced a dose-limiting toxicity (DLT) during coadministration of ketoconazole or esomeprazole, an additional six patients would be enrolled in each arm. If more than one patient experienced a DLT, an additional 12 patients would be enrolled and would receive a lower pazopanib dose. A DLT during the coadministration periods was defined as a grade 4 hematologic toxicity other than lymphopenia, a grade 3 or grade 4 non-hematologic toxicity other than alopecia, nausea, vomiting, or diarrhea in the absence of adequate supportive therapy or any toxicity considered dose-limiting by the investigator. Adverse events were evaluated and graded using the National Cancer Institute Common Technology Criteria for Adverse Events, version 3.0.
The primary endpoints for Arm A were the plasma pazopanib AUC(0–24), maximum observed concentration (C max), and t max after at least 7 consecutive days of administration of pazopanib alone and after 5 days of administration of pazopanib and ketoconazole. The primary endpoints for Arm B were plasma pazopanib AUC(0–24), C max, and t max after at least 7 days of administration of pazopanib alone and after 5 days of administration of pazopanib and esomeprazole. Secondary endpoints included the incidence and severity of AEs, plasma pazopanib concentration 24 h after administration (C 24), pazopanib AUC(0–24), C max, and t max in the presence of ketoconazole or esomeprazole and plasma ketoconazole concentrations.
Pazopanib was administered orally (with a full glass of water) once daily at least 1 h before or 2 h after a meal in the morning. In Arm A, ketoconazole was administered orally once daily before the pazopanib dose in the morning, for a total of 5 days. In Arm B, esomeprazole was administered orally once daily at bedtime, approximately 3 h after the evening meal, for a total of 5 days.
Assessments
Medical history, physical examination, laboratory tests, and clinical chemistry were performed 1 day before the first dose of pazopanib. Vital signs, laboratory tests, and clinical chemistry were also obtained on the day after the final dose of pazopanib and at follow-up (14–28 days after the final dose of pazopanib). Liver function tests and a 12-lead electrocardiogram were repeated on day 7 of period 1. Patients were monitored continuously for AEs.
Pharmacokinetic sampling and statistical analysis
In Arms A and B, on day 7 of period 1 and day 5 of period 2, blood samples for the determination of plasma pazopanib and its metabolites were obtained pre-dose and at 1, 2, 3, 4, 6, 8, and 24 h after pazopanib administration. Plasma samples were analyzed for pazopanib and its metabolites as previously described [5]. Blood samples for the determination of plasma ketoconazole (Arm A) were obtained pre-dose and at 1 and 2 h after pazopanib administration on the last day of period 2. Plasma samples were analyzed for ketoconazole using a validated analytical method based on the protein precipitation, followed by high-performance liquid chromatography/mass spectrometry/mass spectrometry analysis. The lower limit of quantification for ketoconazole was 50 ng/mL using a 50 μL aliquot of human plasma with a higher limit of quantification of 5,000 ng/mL for ketoconazole. Systemic concentrations of esomeprazole were not related to its pharmacodynamic effect on pazopanib; therefore, plasma concentrations of esomeprazole were not quantified.
Pharmacokinetic parameters were calculated by standard non-compartmental methods using WinNonlin (Pharsight Corporation, Mountain View, CA) and summarized descriptively. To evaluate the effect of ketoconazole on the pharmacokinetics of pazopanib, loge-transformed AUC(0–24) and C max were fit to a mixed-effects model with a fixed-effect term for drug treatment and a random effect term for patient. Estimates and 90 % confidence intervals (CIs) were constructed for the difference in least-squares means between treatments. Those results were back-transformed to provide ratios of geometric least-squares means and 90 % CIs for the selected parameters.
The target sample size of 12 evaluable patients per arm was chosen considering the largest within-patient coefficient of variation of 38.5 % from previous pazopanib studies. With a sample size of at least 12 evaluable patients, it was estimated that the precision (i.e., half-width of the 90 % CI on the loge scale) for the treatment difference would be within 32 % of the point estimate for AUC(0–24) and C max.
Results
Patients
Between August 2010 and August 2011, a total of 34 patients were enrolled (Table 1). There were 21 patients treated with pazopanib 400 mg and ketoconazole 400 mg (Arm A), and 13 patients treated with pazopanib 800 mg and esomeprazole 40 mg (Arm B). Overall, the most common malignancies were breast cancer (21 %), colorectal cancer (12 %), uterine cancer (9 %), and ovarian cancer (9 %).
Dose-limiting toxicities
In Arm A, one of the first six patients enrolled experienced a DLT of grade 3 hypertension during period 2. This led to the enrollment of additional patients dosed at 400 mg of pazopanib. Subsequently, a second DLT of grade 3 hypertension occurred during period 2. In addition to the two patients who experienced DLTs, three other patients were not evaluable for pharmacokinetics because of study day vomiting incidents that resulted in dose interruptions. This led to the enrollment of additional patients to obtain evaluable pharmacokinetic data. In Arm B, no DLTs occurred. All 13 patients received pazopanib at 800 mg. There was one patient in Arm B whose pharmacokinetic samples were not evaluable.
Pharmacokinetics
Arm A
Plasma samples for analysis of the pharmacokinetics of pazopanib, and its metabolites were obtained from 21 patients who received pazopanib 400 mg/day and from 16 patients who received pazopanib 400 mg/day plus ketoconazole 400 mg/day. In the presence of ketoconazole, the mean AUC(0–24) increased from 786 to 1,300 μg h/mL and the mean C max rose from 40.7 to 59.2 μg/mL; geometric least-squares mean ratios of these parameters were 1.66 (90 % CI 1.39, 1.99) and 1.45 (90 % CI 1.14, 1.86), respectively (Table 2). The mean concentration–time profiles of pazopanib when administered alone or following concomitant administration of ketoconazole are illustrated in Fig. 1.

Mean (±standard error) plasma concentrations of pazopanib following administration of pazopanib 400 mg for 7 days or coadministration of pazopanib 400 mg plus ketoconazole 400 mg for 5 days (linear scale). Inset semi-log scale
The mean AUC(0–24) and C max for the pazopanib metabolites GSK1268997 and GSK1071306 were lower with concomitant administration of ketoconazole (Table 2). For GSK1268997, AUC(0–24) decreased by 61 % and C max decreased by 68 %; for GSK1071306, AUC(0–24) decreased by 44 % and C max decreased by 43 % (Table 2). Exposure to the pazopanib metabolite GSK1268992, as measured by AUC(0–24) and C max, was unchanged by ketoconazole (Table 2).
Coadministration of ketoconazole 400 mg/day had little effect on the t max of pazopanib (Table 2). Ketoconazole shortened pazopanib t max by 0.45 h (median difference; 90 % CI −1.06, 0.06 h). In contrast, t max for the metabolites of pazopanib was increased (median difference) by more than 9 h [GSK1268992, 9.8 h (90 % CI 7.5, 19.9 h); GSK1268997, 9.4 h (90 % CI 5.5, 11.7 h); GSK1071306, 10.0 h (90 % CI 7.0, 19.0 h)].
The mean plasma concentrations of ketoconazole at 1 and 2 h after administration on day 5 of period 2 in 16 patients were 4.21 μg/mL (range 0.179–10.5 μg/mL) and 4.82 μg/mL (range 0.238–8.4 μg/mL), respectively. These results were consistent with values reported in other studies after administration of ketoconazole 400 mg daily for 4 days [13, 14].
Arm B
Plasma samples for analysis of the pharmacokinetics of pazopanib and its metabolites were obtained from 12 patients who received pazopanib 800 mg/day alone and pazopanib 800 mg/day plus esomeprazole 40 mg/day. In the presence of esomeprazole, the mean AUC(0–24) decreased from 848 to 512 μg h/mL and the mean C max decreased from 48.9 to 28.4 μg/mL; geometric least-squares mean ratios of these parameters were 0.60 (90 % CI 0.52, 0.70) and 0.58 (90 % CI 0.50, 0.67), respectively (Table 3). These data indicate that coadministration of esomeprazole decreased the AUC(0–24) and C max of pazopanib by 40 and 42 %, respectively. The mean concentration–time profiles of pazopanib when administered alone or in combination with esomeprazole are shown in Fig. 2.

Mean (±standard error) plasma concentrations of pazopanib following administration of pazopanib 800 mg for 7 days or coadministration of pazopanib 800 mg plus esomeprazole 40 mg for 5 days (linear scale). Inset semi-log scale
The mean values of AUC(0–24) and C max for the pazopanib metabolites GSK1268992, GSK1268997, and GSK1071306 were lower with concomitant administration of esomeprazole (Table 3). For GSK1268992, both AUC(0–24) and C max decreased by 42 %; for GSK1268997, AUC(0–24) decreased by 48 % and C max decreased by 49 %; for GSK1071306, AUC(0–24) decreased by 30 % and C max decreased by 31 % (Table 3).
Coadministration of esomeprazole increased t max of pazopanib (Table 3); from least-squares analysis, the median difference was 1.07 h (90 % CI –0.10, 2.44 h). The t max for pazopanib metabolite GSK1071306 increased by 1.47 h (median difference, 90 % CI –0.02, 3.75 h). Similar analyses revealed that in the presence of esomeprazole, t max for the metabolites GSK1268992 and GSK1268997 increased slightly (GSK1268992: median difference 0.41 h, 90 % CI −0.20, 1.44 h; GSK1268997: median difference 0.41 h, 90 % CI −0.56, 1.42 h).
Safety
Overall, the most common drug-related AEs were nausea (29 %), vomiting (21 %), hypertension (21 %), decreased appetite (18 %), and diarrhea (15 %) (Table 4). Adverse events were more frequently observed in patients receiving both study drugs than in patients receiving pazopanib only. The majority of AEs in either arm were grade 1 or 2. In Arm A, four patients experienced grade 3 or 4 AEs that were considered drug-related. One patient receiving pazopanib alone experienced grade 3 hypertension that was resolved within 24 h. Among patients who received pazopanib plus ketoconazole, one experienced grade 4 hypokalemia, one experienced grade 3 lymphopenia, and one experienced grade 3 hypertension that led to withdrawal from the trial. An additional patient withdrew from the trial because of grade 3 hypertension, but that event was not considered drug-related. In Arm B, two patients receiving pazopanib and esomeprazole experienced grade 3 hypertension that was considered drug-related. However, both patients had hypertension at baseline and were adequately managed without dose reduction or dose interruption, and the events were not considered DLTs. No serious AEs or deaths occurred during the study. No prolongation of the QTc interval or severe liver toxicity was observed.
Discussion
Potential drug interactions with pazopanib have been explored previously. Administration of pazopanib with a cocktail of CYP450-specific probe drugs (Cooperstown 5 + 1 cocktail) suggested that pazopanib is a weak inhibitor of CYP3A4, its main metabolic enzyme, and had the potential to increase the systemic concentrations of other CYP3A4-metabolized drugs [5]. One of the objectives of the present study was to determine the effect of CYP3A4 inhibition on the pharmacokinetics and safety of pazopanib.
Pharmacokinetic analyses revealed that systemic exposure of pazopanib was increased by the strong CYP3A4 inhibitor ketoconazole. Pazopanib AUC(0–24) and C max increased 66 and 45 %, respectively, following coadministration of pazopanib and ketoconazole, with concomitant decreases in those parameters for the metabolites GSK1268997 and GSK1071306. These results are consistent with an increase in pazopanib exposure due to decreased metabolism in the presence of a CYP3A4 inhibitor. Metabolite GSK1268992 exposure was not changed in the presence of ketoconazole, suggesting that other metabolic pathways are also involved and play a minor role in the systemic clearance of pazopanib.
The recommended therapeutic dose of pazopanib in patients with normal hepatic function is 800 mg once daily. Higher doses were associated with increased toxicity [4]. In the present trial, daily administration of pazopanib 400 mg plus ketoconazole 400 mg led to steady-state systemic exposures (mean AUC(0–24) = 1,300 μg h/mL; mean C max = 59.2 μg/mL) that were comparable to values reported in a study of pazopanib 800 mg once daily in the absence of CYP3A4 inhibition. In that trial, at the recommended therapeutic dose, values for mean AUC(0–24) and mean C max at steady-state were reported to be 1,040 μg h/mL and 58 μg/mL, respectively [5]. Administration of pazopanib at 800 mg in the presence of a strong CYP3A4 inhibitor could therefore lead to increased systemic exposure of pazopanib and increased risk of toxicity. The current prescribing information recommends that coadministration of pazopanib and strong CYP3A4 inhibitors should be avoided. If coadministration is necessary, the dose of pazopanib should be reduced to 400 mg once daily.
Another objective of this trial was to study the effect of elevated gastric pH on the pharmacokinetics of pazopanib. A previous trial revealed that administration of pazopanib with food was associated with an approximately two-fold increase in systemic exposure to the drug [15]. The current prescribing information recommends that pazopanib be administered in the fasted state to minimize day-to-day variations in exposure. In the present study, the protocol to test potential interaction between pazopanib and esomeprazole specified that esomeprazole 40 mg was to be administered in the evening and pazopanib 800 mg was to be administered in the fasted state in the morning. The results of this study demonstrate that at steady-state, systemic exposure to pazopanib was reduced by esomeprazole. Mean AUC(0–24) decreased by 40 % and mean C max decreased by 42 %; steady-state blood levels of all 3 metabolites of pazopanib were similarly decreased. These results suggest that absorption of pazopanib is hindered by elevated fasting gastric pH and is consistent with the observation that the solubility of pazopanib is greater in a more acidic environment [15].
Results from a phase I trial suggested that the clinical efficacy of pazopanib is related to a threshold steady-state trough concentration (C 24) of at least 15 μg/mL [4]. In the present study, the mean values for C24 in patients receiving pazopanib 800 mg once daily in the absence of esomeprazole were 27.2 μg/mL (Table 3). In the presence of esomeprazole, the mean C 24 of pazopanib dropped to 17.3 μg/mL (Table 3). Since the latter value is close to the threshold concentration for therapeutic efficacy, and there can be substantial between-patient variability in exposure at the same dose level, coadministration of esomeprazole may create the potential for subtherapeutic exposure of pazopanib in some patients. If pazopanib and esomeprazole must be used together, care should be taken to administer pazopanib when gastric pH is expected to be at a minimum. Gastric pH has been observed to follow a circadian pattern, with pH tending to fall during the nighttime fasting period and then rise in the morning hours of 6–8 a.m. [16]. A study that examined gastric pH following an evening dose of esomeprazole 40 mg found that the value of gastric pH reached approximately 6.0 during the morning peak just before breakfast and was at a minimum of approximately 3.0 just before dinner [17]. To avoid reduced absorption and subtherapeutic blood levels, the dosing schedule of pazopanib therefore should be tailored to match a minimum in gastric pH in patients receiving esomeprazole or other drugs that reduce gastric acidity.
The interactions between pazopanib and the other study drugs appeared to have little effect on safety, as pazopanib was well tolerated in this study. Most AEs were grade 1 or 2 in either arm, and no new or unexpected AEs were reported. Hypertension, an AE known to be associated with pazopanib therapy, was observed in no more than 15 % of patients in either study arm; all events of hypertension were well managed. More AEs were observed during the second phase of each study arm, when pazopanib was being coadministered with either ketoconazole or esomeprazole. However, the design of the study does not permit a rigorous evaluation of whether the increase in AEs was caused by the increased time on pazopanib therapy or by drug–drug interactions.
Conclusion
Administration of pazopanib in the presence of a CYP3A4 inhibitor, such as ketoconazole, increased systemic exposure to pazopanib. Concomitant use of pazopanib with a strong inhibitor of CYP3A4 should be avoided. A reduced dose of pazopanib 400 mg daily should be given if concomitant administration with a CYP34A inhibitor is necessary. There was a reduction in the absorption of pazopanib when coadministered with esomeprazole at the 40 mg/day dose. If pazopanib and proton pump inhibitors are used concomitantly, the dosing pattern should avoid administration of the proton pump inhibitor at bedtime and administration of pazopanib the following morning, when gastric pH is maximal. Consideration should be given to changing pazopanib administration at a time when gastric pH is expected to be low, such as late evening.
References
Harris PA, Boloor A, Cheung M et al (2008) Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-b enzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem 51:4632–4640
Sternberg CN, Davis ID, Mardiak J et al (2010) Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 28:1061–1068
van der Graaf WT, Blay JY, Chawla SP et al (2012) Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379:1879–1886
Hurwitz HI, Dowlati A, Saini S, Savage S, Suttle AB, Gibson DM, Hodge JP, Merkle EM, Pandite L (2009) Phase I trial of pazopanib in patients with advanced cancer. Clin Cancer Res 15:4220–4227
Goh BC, Reddy NJ, Dandamudi UB et al (2010) An evaluation of the drug interaction potential of pazopanib, an oral vascular endothelial growth factor receptor tyrosine kinase inhibitor, using a modified Cooperstown 5 + 1 cocktail in patients with advanced solid tumors. Clin Pharmacol Ther 88:652–659
Abbas R, Hug BA, Leister C, Burns J, Sonnichsen D (2011) Pharmacokinetics of oral neratinib during co-administration of ketoconazole in healthy subjects. Br J Clin Pharmacol 71:522–527
Dutreix C, Peng B, Mehring G, Hayes M, Capdeville R, Pokorny R, Seiberling M (2004) Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjects. Cancer Chemother Pharmacol 54:290–294
Johnson FM, Agrawal S, Burris H et al (2010) Phase 1 pharmacokinetic and drug-interaction study of dasatinib in patients with advanced solid tumors. Cancer 116:1582–1591
Di Gion P, Kanefendt F, Lindauer A, Scheffler M, Doroshyenko O, Fuhr U, Wolf J, Jaehde U (2011) Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on pyrimidines, pyridines and pyrroles. Clin Pharmacokinet 50:551–603
Miner P Jr, Katz PO, Chen Y, Sostek M (2003) Gastric acid control with esomeprazole, lansoprazole, omeprazole, pantoprazole, and rabeprazole: a five-way crossover study. Am J Gastroenterol 98:2616–2620
Yin OQ, Gallagher N, Fischer D, Demirhan E, Zhou W, Golor G, Schran H (2010) Effect of the proton pump inhibitor esomeprazole on the oral absorption and pharmacokinetics of nilotinib. J Clin Pharmacol 50:960–967
Kapadia S, Hapani S, Wu S (2011) Risk of high-grade liver toxicity with pazopanib in patients with cancer: a meta-analysis. J Clin Oncol 29(suppl):abstract 4595
Olkkola KT, Backman JT, Neuvonen PJ (1994) Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther 55:481–485
Bjornsson TD, Callaghan JT, Einolf HJ et al (2003) The conduct of in vitro and in vivo drug–drug interaction studies: a Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos 31:815–832
Heath EI, Chiorean EG, Sweeney CJ et al (2010) A phase I study of the pharmacokinetic and safety profiles of oral pazopanib with a high-fat or low-fat meal in patients with advanced solid tumors. Clin Pharmacol Ther 88:818–823
Warrington S, Baisley K, Boyce M, Tejura B, Morocutti A, Miller N (2002) Effects of rabeprazole, 20 mg, or esomeprazole, 20 mg, on 24-h intragastric pH and serum gastrin in healthy subjects. Aliment Pharmacol Ther 16:1301–1307
Wilder-Smith C, Rohss K, Bokelund Singh S, Sagar M, Nagy P (2010) The effects of dose and timing of esomeprazole administration on 24-h, daytime and night-time acid inhibition in healthy volunteers. Aliment Pharmacol Ther 32:1249–1256
Acknowledgments
Financial support for this study and for medical editorial assistance was provided by Glaxo SmithKline Pharmaceuticals, Philadelphia, Pennsylvania. We thank William Sinkins, PhD, ProEd Communications, Inc., for his medical editorial assistance with this manuscript.
Conflict of interest
Authors Suttle and Tada are GlaxoSmithKline employees and stockholders. Author Botbyl’s company, Provonix, provides data services to GlaxoSmithKline. Author Tan has received research funding from GlaxoSmithKline. All other authors report no potential conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Clinical Trials Registration Number: NCT01205230.
Rights and permissions
About this article
Cite this article
Tan, A.R., Gibbon, D.G., Stein, M.N. et al. Effects of ketoconazole and esomeprazole on the pharmacokinetics of pazopanib in patients with solid tumors. Cancer Chemother Pharmacol 71, 1635–1643 (2013). https://doi.org/10.1007/s00280-013-2164-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-013-2164-3