
Abstract
Purpose
The histone deacetylase inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA) enhances taxol-induced antitumor effects against some human cancer cells. The aim of this study is to investigate whether SAHA can enhance taxol-induced cell death against human breast cancer cells and to illustrate the mechanism in detail.
Methods
A panel of eight human breast cancer cell lines and an immortalized human breast epithelial cell line were used to determine the inhibitory effects of SAHA, taxol, or their combination by MTT assay. The effects of SAHA with or without taxol on cell cycle distributions, apoptosis, and protein expressions were also examined. The inhibitory effects on tumor growth were characterized in vivo in BALB/c nude mice bearing a breast cancer xenograft model.
Results
Taxol-resistant and multi-resistant breast cancer cells were as sensitive to SAHA as taxol-sensitive breast cancer cells. A dose-dependent synergistic growth inhibition was found in all the tested breast cancer cell lines treated with the SAHA/taxol combinations. The synergetic effect was also observed in the in vivo xenograft tumor model. The cell cycle analysis and apoptosis assay showed that the synergistic effects resulted from enhanced G2/M arrest and apoptosis.
Conclusions
SAHA increased the anti-tumor effects of taxol in breast cancer in vitro and in vivo. The combination of SAHA and taxol may have therapeutic potential in the treatment of breast cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Histone deacetylases (HDACs) and histone acetylases (HATs) are enzymes responsible for deacetylating and acetylating the amino-terminal lysine residues of histones. HDACs remove acetyl groups from core histones, thereby compacting the chromatin structure and suppressing gene expression [1]. The activity of these enzymes is not restricted to histones, and a growing number of non-histone proteins have been identified as HDACs/HATs targets [2]. As they are involved in a broad range of cell functions, HDACs are considered an important class of cancer targets, and HDAC inhibitors (HDACi) are becoming a promising new class of anticancer drugs. Several HDACis have already been in preclinical and clinical development [3], among which suberoylanilide hydroxamic acid (SAHA, generic name vorinostat) showed single-agent anti-proliferative activity against a variety of preclinical tumor models [4–7] and is now applied in the clinical treatment of cutaneous T-cell lymphomas [8]. Many studies have shown that SAHA induces cell cycle arrest, differentiation, and apoptosis both in vitro and in vivo [9].
Taxol is one of the most extensively used anticancer agents, with clinical effects against a wide range of cancers. Taxol stabilizes microtubules, thus preventing cell proliferation at the metaphase/anaphase boundary [10]. However, the success of taxol in clinical treatment was tempered by drug resistance, which severely limited the effects of chemotherapy. Although the exact mechanism remains unclear, the drug resistance is considered to be mediated by altered drug uptake, variations in tubulin structure, and evasion of apoptotic pathways [11].
As HDACi show promising results as anti-cancer reagents without obvious toxicity against normal cells, their combination with other reagents may improve the overall efficiency and breadth of application. HDACi have already been shown to function synergistically in vitro with structurally and functionally diverse chemical compounds [3]. Recently, several studies have shown that SAHA enhances the growth inhibitory effect induced by taxol against lung cancer cells, ovarian cancer cells, and endometrial cancer cells [12–15]. However, whether similar effects would occur against breast cancer cells in vitro and in vivo has not been investigated. In this study, SAHA was proven to enhance taxol-induced cell death in human breast cancer cells, and a potential mechanism for this synergetic effect was also proposed.
Materials and methods
Cell lines and cell culture
All human breast cancer cell lines used in this study were purchased from the Shanghai Cell Bank, the Institute of Cell Biology, China Academy of Sciences (Shanghai, China). All cell lines were maintained in a humidified atmosphere containing 5% CO2 at 37°C in different media supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. MDA-MB-231, MCF-7, MCF-7/ADR, and SKBR3 were cultured in RPMI-1640 medium. BT474 and MDA-MB-453 cells were cultured in DMEM. T-47D cells were cultured in DMEM/F12. MDA-MB-435s cells were cultured in L-15 medium. An immortalized human mammary epithelial cell line MCF10A was purchased from the American Type Culture Collection and cultured in DMEM supplemented with 5% horse serum, 20 ng/ml of epidermal growth factor, 0.5 μg/ml of hydrocortisone, 100 ng/ml of cholera toxin, 10 μg/ml of insulin, and penicillin/streptomycin.
Inhibition of cell growth in vitro
The cells were seeded into 96-well plates and incubated overnight. The cells were then treated for 72 h with varying concentrations of SAHA (Sigma, USA), taxol (Bristol-Myers Squibb, USA), and a combination of SAHA and taxol at a molar ratio of 1:2. 20 μl of 3-(4,5 dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide (MTT, Sigma, USA) solution (5 mg/ml in PBS) were added to each well and incubated for 4 h at 37°C. After the removal of the medium, MTT formazan was dissolved in 150 μl dimethyl sulfoxide (DMSO) and monitored using a microplate reader at a wavelength of 570 nM.
Drug synergetic effect studies
The combination effects of SAHA and taxol were subjected to median effect analysis as previously described by Chou and Talalay [16]. By combining SAHA and taxol at a fixed molar ratio of doses [at the ratio of their half-maximal inhibitory concentrations (IC50s)], numerous combined effects of growth inhibition were obtained and analyzed using Calcusyn software (Biosoft, Cambridge, UK). For each combined effect of growth inhibition or fraction affected (Fa), a combination index (CI) was generated. CI < 1, CI = 1, and CI > 1 indicate synergism, additive effect, and antagonism, respectively [17]. The dose reduction index (DRI) represents the measure of how much the dose of each drug in a synergistic combination may be reduced at a given effect level compared with the doses of each drug alone.
Cell cycle analysis by flow cytometry
The cells were trypsinized, fixed in ice-cold 70% ethanol, and then stored at −20°C. Prior to analysis, the samples were washed twice in phosphate-buffered saline (PBS) and resuspended in a solution of propidium iodide (50 mg/ml) and RNase A (0.5 mg/ml) in PBS for 30 min in the dark. Data collected from each 10,000-cell sample was analyzed by flow cytometry (Becton–Dickinson Co., USA).
Annexin V/Propidium iodide assay
Annexin V/Propidium iodide (PI) binding assay was employed to determine the viable, early apoptotic cells. Following the recommended protocols of the Annexin V-FITC kit (BD Pharmingen, USA), the cells were seeded at 4 × 105 cells/ml per well in 6-well plates. After treatment with SAHA, taxol alone, or both, the MDA-MB-231 cells were harvested and washed twice with ice-cold PBS and resuspended in 100 μl of binding buffer. A total of 5 μl of Annexin V-FITC and 10 μl of PI were added, and the mixture was incubated for 30 min in the dark. Finally, 400 μl of binding buffer was added to the cells, and the mixture was analyzed with a flow cytometer (Becton–Dickinson Co., USA).
Terminal deoxynucleotidyl transferase dNTP nick end labeling (TUNEL) staining
Apoptotic cells were detected in representative sections of tumor tissue through staining with terminal deoxynucleotidyl transferase dNTP nick end labeling (TUNEL) following the manufacturer’s instructions (FragEL™ DNA Fragmentation Detection Kit, Fluoresent-TdT enzyme, Calbiochem, Germany).
Western blot analysis
The cells were trypsinized, washed with PBS, and then lysed with buffer containing 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1 mM dithiothreitol, 1% Nonidet P-40, 0.1% SDS, protease inhibitors (1 mM PMSF, 5 mg/ml aprotinin, 5 mg/ml leupeptin and 5 mg/ml pepstatin), and phosphatase inhibitors (20 mM β-glycerophosphate, 50 mM NaF, and 1 mM Na3VO4). The lysates were incubated at 4°C for 20 min and centrifuged at 12,000×g for 15 min. Equal amounts of lysate (40 μg) were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to polyvinylidene difluoride membrane (Millipore, USA). The membranes were blocked in 5% non-fat skim milk/TBST [20 mM Tris–HCl (pH 7.4), 150 mM NaCl, and 0.1% Tween-20] at room temperature for 2 h and detected with primary antibodies at room temperature for 2 h. The membranes were then blotted for 1 h at room temperature with an appropriate horseradish peroxidase-linked secondary antibody, followed by enhanced chemiluminescence Western blot detection reagents (Amersham Pharmacia Biotech, USA). The primary antibody cyclin D1, cyclin B1, p21, caspase 3, Bax, acetylated α-tubulin, actin, and the secondary antibody were purchased from Santa Cruz Biotechnology (USA); anti-acetyl-Histone H3 was purchased from Millipore (USA).
Inhibition of tumor growth in vivo
The research protocol was in accordance with the institutional guidelines of the Animal Care and Use Committee of Shandong University. The animals were housed under pathogen-free conditions. Female BALB/c (nu/nu) mice (20 ± 2 g, 4–6 weeks old) were purchased from the Animal Center of the China Academy of Medical Sciences (Beijing, China). The breast cancer MDA-MB-231 cells, 5.0 × 106, suspended in 100 μl PBS, were subcutaneously inoculated into the lower right flank of the nude mice. When the tumors were 100–150 mm3, the mice were divided randomly into four groups (n = 6 in each group). The control group received the solvent (DMSO) only. The treatment groups received SAHA (50 mg/kg), taxol (7.5 mg/kg), or a combination of SAHA (50 mg/kg) and taxol (7.5 mg/kg). The mice receiving DMSO or SAHA treatment were injected intraperitoneally with 100 μl of DMSO or SAHA (diluted in 100 μl DMSO) on days two, three, four, six, and seven of each week. The mice receiving taxol treatment were injected intraperitoneally with 200 μl of taxol on days one and five of each week. The mice were treated for 3 weeks. The diameter of the tumor was measured twice a week with a caliper. Tumor volume was calculated with the following formula: v = ab 2/2, where a and b are the long diameter and the perpendicular short diameter of the tumor, respectively. At the end of the experiment, the tumors were harvested, and the tumor weights were determined.
Statistical analysis
All quantitative data were subject to ANOVA to determine if there were significant differences between groups. For data groups satisfying the ANOVA criteria (P < 0.05), individual comparisons were conducted using the Student’s t-test; P ≤ 0.05 was considered significant.
Results
SAHA enhanced taxol-induced cell death in breast cancer cells
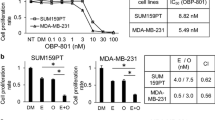
Some studies have shown that the growth inhibition of breast cancer cells by SAHA is not dependent on p53, estrogen receptor (ER), human epidermal growth factor receptor 2 (HER2), and retinoblastoma (RB) expression. To determine whether SAHA could enhance the growth inhibition of taxol in breast cancer cells, an MTT assay was performed using different breast cancer cell lines and an immortalized human breast epithelial cell line MCF10A with various protein expression levels of p53, ER, HER2, and RB. As seen in Fig. 1a and Table 1, all breast cancer cells were sensitive to SAHA, with IC50 between 1.90 and 5.10 μM, whereas MCF10A showed an IC50 value of 10.75 ± 2.21 μM. Multi-resistant MCF-7/ADR cells also showed higher resistance to SAHA than the other cancer cell lines, with an IC50 of 5.10 ± 0.21 μM. These results prove that the growth inhibition of breast cancer cells by SAHA has a broad spectrum and is not dependent on p53, ER, HER2, and RB expression. Furthermore, the cancer cells were more sensitive to SAHA than the immortalized MCF10A cells. For taxol, both MCF-7/ADR and SKBR3 cells showed resistance to taxol, whereas all other cells were similarly sensitive (Fig. 1b).

Growth inhibitory effects of SAHA and taxol in breast cancer cell lines. The cells were exposed to either SAHA (a) or taxol (b) for 72 h. The growth inhibitory effects were determined by MTT assay. The results were derived from three independent experiments performed in triplicate
Based on the IC50 values of SAHA or taxol alone on the different cancer cells, the combined treatments of SAHA with taxol were initiated simultaneously at equipotent doses of the two drugs (at the ratio of their IC50s) to test whether there were synergistic effects against all breast cancer cells and MCF10A cells. The combination effects, such as median effect, dose effect, and CI, were obtained using CalcuSyn software. The CI-Fa curve in Fig. 2a–i indicates that the SAHA and taxol combinations produced a synergistic effect against all the tested cells. However, these synergisms depended on the concentrations of SAHA and taxol. For MCF10A cells, synergistic growth inhibitory effect was observed in all the doses used in the MTT assay. For the taxol-resistant breast cancer cells SKBR3 and MCF-7/ADR, the synergistic effects were observed when the concentration of taxol was higher than 4.0 nM and SAHA was higher than 2.0 μM. For non-resistant cell lines, such as MDA-MB-231, MCF-7, MDA-MB-453, MDA-MB-435s, T-47D, and BT474, synergism was observed with low SAHA and taxol concentrations (C[taxol] < 4.0 nM and C[SAHA] < 2.0 μM). CI and dose reduction index (DRI) values for all the breast cancer cells are summarized in Table 1. Both SAHA and taxol had favorable DRIs. At IC50 effect levels, the doses of SAHA and taxol decreased in varying degrees, indicating that the combination treatment of SAHA with taxol may lead to reduced toxicity in chemotherapy.

Synergistic growth inhibitory effects of SAHA and taxol on breast cancer cell lines. CI/fractional effect curves show the CI versus the fraction of cells affected/killed by the combination of SAHA and taxol for the different breast cancer cell lines (a–i). Dose–effect plots (j) and median-effect plots (k) show the interaction between SAHA and taxol on cell growth in MDA-MB-231 breast cancer cells. The cells were simultaneously exposed to SAHA and taxol alone or combined at a fixed ratio following a gradient of concentrations. An MTT assay was performed after treatment for 72 h. Combination analysis was done using the method described by Chou and Talalay (see Materials and methods). For these curves, CI < 1.0 indicates a synergistic interaction and CI > 1 signifies antagonistic drug effects. A straight line at the CI = 1 represents the additive effect from both drugs. Representative experiments were carried out at least three times for each cell line
SAHA increased taxol-induced G2/M arrest in breast cancer MDA-MB-231 cells
As similar growth inhibitory effects were obtained from MDA-MB-231, MCF-7, MDA-MB-453, MDA-MB-435s, T-47D, and BT474 cells after treatment with SAHA, taxol or a combination of the two (Fig. 2), MDA-MB-231 cells were selected for the subsequent studies focused on indentifying the mechanism underlying how SAHA enhanced taxol-induced growth inhibition in breast cancer cells.
Cell cycle analysis was used to identify the cell cycle distributions of the MDA-MB-231 cells after exposure to either SAHA, taxol, or the SAHA/taxol combination for 24 h. The results (Fig. 3a) revealed that taxol induced G2/M cell cycle arrest in a dose-dependent manner. SAHA induced G1/G0 arrest at low concentrations and G2/M arrest at high concentrations. The cells treated with a combination of SAHA (0.5 or 2.0 μM) and taxol (1.0 or 4.0 nM) arrested more in the G2/M phase than those treated with either drug alone at the same concentration. Compared with the 8.0 μM SAHA treatment, the treatment with 8.0 μM SAHA combined with different concentrations of taxol had no significant changes in cell cycle distribution. Similar results were also observed with 16 nM taxol combined with various concentrations of SAHA, which induced the same cell cycle distribution pattern as 16 nM taxol alone (Fig. 3a).

Effects of SAHA and taxol on cell cycle distributions (a) and apoptosis (b) in MDA-MB-231 breast cancer cells. The cells were treated with SAHA, taxol, or a combination of SAHA and taxol for 24 h and then analyzed for cell cycle distributions and apoptosis. Representative experiments were carried out at least three times. *P < 0.05, combined treatment of SAHA with taxol versus control, SAHA, or taxol alone
SAHA potentiated taxol-induced apoptosis
Annexin V-FITC/PI analysis was used to examine the percentage of apoptosis in the MDA-MB-231 cells following treatment with SAHA, taxol, or their combination for 24 h. SAHA or taxol alone induced apoptosis in a dose-dependent manner (data not shown). As shown in Fig. 3b, the combination of SAHA with taxol resulted in significant increases in the percentage of apoptotic cells, suggesting that SAHA enhanced taxol-induced apoptosis.
SAHA/taxol treatments changed the expression of cell cycle and apoptosis-associated proteins in MDA-MB-231 cells
To explore the mechanism of the growth inhibition effects of SAHA with or without taxol in breast cancer cells, the expression of proteins associated with the cell cycle and apoptosis were further examined. After being exposed to SAHA at different concentrations for 24 h, the Western blot of MDA-MB-231 cells showed that acetylated histones H3 increased after treatment with 0.5, 2.0, and 8.0 μM SAHA. SAHA reduced the expression of cyclins D1 and B1 and upregulated p21 expression. Both caspase 3 and Bax, which are pro-apoptosis-related proteins, were also elevated with the treatment of SAHA (Fig. 4a). When the MDA-MB-231 cells were treated with SAHA (2.0 μM) combined with taxol (4.0 nM), the expression of the acetylated histones H3 increased similar to the cells treated with SAHA alone; however, taxol alone did not affect the level of the acetylated histones H3 in the cells (Fig. 4a, b). In contrast, the combined treatment significantly increased the expression of p21, caspase 3, and Bax higher than either SAHA or taxol treatment alone (Fig. 4a, b). 2.0 μM SAHA alone had little effect on the expression of cyclin D1 and reversed the reduction of cyclin D1 by taxol. The decreased cyclin B1 expression may be related to the G2/M arrest induced by SAHA or the combined SAHA/taxol treatment. Taxol interferes with microtubule depolymerization and results in the accumulation of acetylated α-tubulin and stabilized microtubule structures, which disrupts the alignment of chromosomes during mitosis and eventually leads to apoptosis. Interestingly, α-tubulin is a non-histone substrate for HDAC enzymes [17]. At least one member of the HDAC family, HDAC-6, is able to remove the acetyl group from tubulin [18]. As tubulin deacetylation is associated with microtubule depolymerization, the accumulation of acetylated tubulin following treatment with SAHA may promote microtubule stabilization. To determine whether SAHA and taxol co-regulate microtubule stability could account for the increased growth inhibition, acetylated tubulin expression was also evaluated. When the MDA-MB-231 cells were exposed to SAHA or taxol, a modest increase in acetylated tubulin was observed. In contrast, a significant increase in the expression of acetylated tubulin was observed when SAHA and taxol were given simultaneously (Fig. 4a, b).

Effects of SAHA and taxol on the expression of apoptosis and cell cycle regulation-related proteins in MDA-MB-231 breast cancer cells. a A result of a Western blot; b Protein expressions induced by combined treatment were quantitated by densitometry and normalized against that of β-actin. Bars in the charts represent means ± SD of three independent experiments. The cells were treated with SAHA, taxol, and the combination of these two drugs for 24 h and were then harvested for Western blot analysis. Ac-H3 represents acetylated Histone H3; Ac-tubulin represents acetylated α-tubulin
SAHA inhibited breast tumor growth and enhanced the in vivo anti-tumor effect of taxol
To determine further the antitumor effects of SAHA and the SAHA/taxol combination, athymic mice were treated with vehicle, SAHA, taxol, or SAHA/taxol combination after the taxol-sensitive MDA-MB-231 xenograft tumor was visible. The growth of the tumors was monitored twice a week, and the volume was calculated by the formula described in the Materials and Methods. The tumors from mice treated by SAHA/taxol combination treatment clearly had smaller sizes and weights than those either from the SAHA or taxol treatment groups (Fig. 5a, b). Compared with the treatment in the control group, SAHA (50 mg/kg), taxol (7.5 mg/kg), and the combined treatment reduced the tumor volume of the MDA-MB-231 xenografts by 31.9, 19.5 and 46.0%, respectively (Fig. 5c). TUNEL assay was used to assess apoptosis in paraffin-embedded tumor sections. Tumors from mice that received the combined treatment showed a larger proportion of cells with TUNEL-positive nuclei than tumors from mice that received the vehicle, taxol, or SAHA treatment alone (Fig. 5d).

Anti-tumor effects of SAHA and taxol on the mice xenograft model. The nude mice bearing breast cancer MDA-MB-231 cells were treated as described. a Mean tumor weights for each group at the end of the treatment; b Imaging of the tumors dissected from xenograft mice at the end of the treatment; c Tumor size growth curves during the treatment calculating the volume size of individual tumors; and d TUNEL detection of apoptosis in tumors following drug treatment. The original magnification was ×400. *P < 0.05, combined treatment of SAHA with taxol versus control, SAHA, or taxol alone
The body weight and survival ratio of the treated mice from each treatment group were also monitored to evaluate the long-term toxicity of SAHA and the combination treatment. There were no significant body weight losses for all treated mice. These data suggest that, compared with the SAHA and taxol single treatments, the combination of SAHA with taxol significantly enhanced the anti-tumor effects without increasing the toxicity.
Discussion
At a steady fed-state, oral administration of 400 mg/m2 SAHA in patients resulted in a mean area under the concentration–time curve (AUC) of 6.0 ± 2.0 μM hour and maximum concentration (Cmax) of 1.2 ± 0.53 μM [19]. Correspondingly, at 135 mg taxol administered by 3 h infusion resulted in an AUC of 9.4–13.1 μM hour and Cmax of 2.5–3.9 μM [20–24]. In this report, the concentrations of SAHA and taxol used in the in vitro study was equal to or lower than the plasma levels in patients treated with SAHA or taxol.
SAHA is a small molecule inhibitor of class I and II HDAC enzymes [25]. While affecting acetylation, SAHA interferes with the cell cycle, apoptosis, and differentiation of human cancer cells. In previous studies, SAHA inhibited MCF-7, MDA-MB-231, MDA-MB-435, and SKBR3 breast cancer cell lines by inducing G1 and G2/M arrest as well as apoptosis [26]. In HER2-overexpressing breast cancer cell lines, in addition to the dose-dependent facilitation of apoptosis, SAHA induced the acetylation of hsp90, leading to the dissociation of HER2 from the chaperone molecule and resulting in polyubiquitylation and degradation of HER2 [27]. SAHA treatment resulted not only in the re-expression of ER but also in the inhibition of epidermal growth factor receptor expression in ER-negative human breast cancer MDA-MB-231 cells [28]. In this study, the inhibitory effects of SAHA, with or without taxol, was observed in an immortalized human breast epithelial cell line MCF10A and in a panel of eight breast cancer cell lines, including multi-resistant MCF-7/ADR cells. The protein expressions of p53, ER, HER2, and RB were varied in these breast cancer cell lines. Concurring with other researchers [26, 29], our study showed that SAHA inhibited cell growth by inducing apoptosis and altering cell cycle distributions. As shown in Table 1, the sensitivity of breast cancer cells to SAHA was not dependent on the expression of p53, ER, HER2, and RB. HER2-overexpressing SKBR3 cells and multi-resistant MCF-7/ADR cells were indeed resistant to taxol; however, they were sensitive to SAHA, with IC50 values of 2.86 μM (95% CI: 1.80–4.53 μM) and 5.10 μM (95% CI: 3.29–7.92 μM), respectively. These data suggest that SAHA had potent growth inhibitory effect in taxol-resistant breast cancer cells and in multi-resistant breast cancer cells. These results are consistent with the previous findings in which SAHA was sensitive to taxol-resistant ovarian cancer cells, and SAHA was shown to induce cell death in P-glycoprotein–overexpressing T-cell leukemia and colon carcinoma cells [30–32]. The result raises the possibility that combined treatment with SAHA and taxol may improve the antitumor effects on taxol-resistant breast cancer. However, only the antitumor effect of the combined treatment of SAHA and taxol in nude mice bearing a taxol-sensitive MDA-MB-231 xenograft was explored in this study. Further investigation on the combined treatment in nude mice bearing taxol-resistant breast cancer xenografts is worthwhile.
Due to the potential antitumor effect of SAHA, it was evaluated alone and in combination with targeting therapeutic or chemotherapeutic agents in preclinical studies and in clinical trials [33, 34]. In drug combination studies, there are different ways to combine the drugs. In this study, the diagonal constant ratio combination design proposed by Chou and Talalay [16] and Chou [35] was used. The diagonal constant ratio combination design can greatly reduce the number of data points required and can still achieve the maximal amount of useful information on the combinations, thus increasing the cost-effectiveness of experimentation. In an early-stage study, the constant combination ratio experiment should be carried out at an equipotency ratio [e.g., (IC50)1/(IC50)2 ratio] to equalize roughly the contributions of the effects of each drug to the combination. In this study, after each IC50 of SAHA and taxol was obtained, the combined treatment was initiated at equipotent doses of the two drugs following their IC50 ratios with serial dilutions for several doses. Using the design, the addition of SAHA to taxol resulted in a dramatic decrease in IC50 values for taxol and also a significantly greater growth inhibition than with either SAHA or taxol alone in eight different breast cancer cell lines and MCF10A cells. Interestingly, the synergistic growth inhibition was dependent on the concentration of SAHA and taxol, as well as the type of cells being treated. Synergism was also observed in immortalized MCF10A cells independent of the SAHA or taxol concentrations. In MDA-MB-231, MCF-7, MDA-MB-453, MDA-MB-435s, T-47D, and BT474 cells, synergism was found at low SAHA and taxol concentrations. However, in taxol-resistant SKBR3 and MCF-7/ADR cells, synergism was found at high concentrations of SAHA and taxol (Fig. 2). The reason for the dependence of the synergism on the concentrations of SAHA and taxol may be related to the mechanism of action of taxol. Torres and Horwitz [36] showed that the mechanism by which taxol induces cell death is dose-dependent. At low concentrations, taxol induces cell death primarily through its microtubule stabilizing effect, leading to G2/M arrest and p21 and p53 protein induction. In contrast, at higher concentrations, taxol induces cell death through a Raf-1 dependent manner. However, further investigations on the involved mechanisms are still needed.
The ability of SAHA to increase the growth inhibition of taxol in breast cancer cells was consistent with prior results obtained using lung cancer, ovarian cancer, and thyroid cancer cells [12–14, 37]. A study in lung cancer cells also found that SAHA potentiated taxol activity at lower concentrations but not at higher concentrations of taxol [12].
The enhanced growth inhibition of SAHA with taxol may be associated with cell cycle distributions induced by both agents. Cell cycle analysis demonstrated that SAHA induced G1/G0 arrest at low concentrations and G2/M arrest at high concentrations, and that taxol induced G2/M arrest in MDA-MB-231 cells. Low concentrations of SAHA elevated the percentage of G2/M phase cells induced by low concentrations of taxol. As either SAHA or taxol alone at high concentrations sharply caused G2/M arrest, high concentrations of SAHA with different concentrations of taxol or high concentrations of taxol with different concentrations of SAHA did not show significant changes in cell cycle distributions compared with SAHA or taxol at high concentrations. The increased G2/M phase cells induced by SAHA with taxol at low concentrations resulted from the upregulation of p21 and downregulation of cyclin B1. As shown in Fig 4, SAHA augmented the expression of acetylated histones H3 and p21, and decreased the expression of cyclins D1 and B1. The induction of p21 is an important mechanism by which taxol induces apoptosis [38]. Similarly, HDACis, such as SAHA, are also known to induce p21, which interacts with Rho to promote the accumulation of stabilized, detyrosinated microtubules [39, 40]. Although p21 is commonly associated with the G1 check point, its association with G2/M cell cycle arrest has also been demonstrated [41]. Cyclin B1 plays a critical role in regulating cell-cycle progression from G2 through M phase (including exit from M phase). In this study, enhanced G2/M cell cycle arrest could also be associated with the decreased cyclin B1 expression induced in MDA-MB-231 cells by the combined SAHA with taxol treatment.
One of the most important mechanisms by which taxol inhibit tumor growth is by microtubule stabilization, which occurs through its action on tubulin acetylation. Our data showed that when added to taxol, SAHA caused a marked increase in the expression of acetylated tubulin. The high expression of acetylated tubulin could result in enhanced apoptosis. As shown by Annexin V-FITC/PI analysis and TUNEL staining, more apoptotic cells were found after combined treatment of SAHA with taxol in vitro and in vivo. The enhanced apoptosis was also verified by the increased expression of caspase 3 and Bax after the cells were treated with the combination of SAHA and taxol.
In conclusion, SAHA had potent growth inhibitory effects in breast cancer cells, even in taxol-resistant or multi-resistant breast cancer cells. The synergistic antitumor activity of SAHA with taxol was dependent on concentrations of both drugs and resulted from enhanced G2/M arrest and apoptosis. The combination of SAHA and taxol may be useful in the treatment of breast cancer.
Abbreviations
- CI:
-
Combination index
- DRI:
-
Dose reduction index
References
Glaser KB (2007) HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol 74:659–671
Zhou Q, Chaerkady R, Shaw PG, Kensler TW, Pandey A, Davidson NE (2010) Screening for therapeutic targets of vorinostat by SILAC-based proteomic analysis in human breast cancer cells. Proteomics 10:1029–1039
Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5:769–784
Neureiter D, Zopf S, Leu T, Dietze O, Hauser-Kronberger C, Hahn EG, Herold C, Ocker M (2007) Apoptosis, proliferation and differentiation patterns are influenced by Zebularine and SAHA in pancreatic cancer models. Scand J Gastroenterol 42:103–116
Lindemann RK, Newbold A, Whitecross KF, Cluse LA, Frew AJ, Ellis L, Williams S, Wiegmans AP, Dear AE, Scott CL, Pellegrini M, Wei A, Richon VM, Marks PA, Lowe SW, Smyth MJ, Johnstone RW (2007) Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci USA 104:8071–8076
Cohen LA, Marks PA, Rifkind RA, Amin S, Desai D, Pittman B, Richon VM (2002) Suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor, suppresses the growth of carcinogen-induced mammary tumors. Anticancer Res 22:1497–1504
Imre G, Gekeler V, Leja A, Beckers T, Boehm M (2006) Histone deacetylase inhibitors suppress the inducibility of nuclear factorkappaB by tumor necrosis factor-alpha receptor-1 down-regulation. Cancer Res 66:5409–5418
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12:1247–1252
Frew AJ, Johnstone RW, Bolden JE (2009) Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett 280:123–133
Mo Y, Gan Y, Song S, Johnston J, Xiao X, Wientjes MG, Au JL (2003) Simultaneous targeting of telomeres and telomerase as a cancer therapeutic approach. Cancer Res 63:579–585
Kavallaris M (2010) Microtubules resistance to tubulin-binding agents. Nat Rev Cancer 10:194–204
Owonikoko TK, Ramalingam SS, Kanterewicz B, Balius TE, Belani CP, Hershberger PA (2010) Vorinostat increases carboplatin and paclitaxel activity in non-small-cell lung cancer cells. Int J Cancer 126:743–755
Dietrich CS 3rd, Greenberg VL, DeSimone CP, Modesitt SC, Van Nagell JR, Craven R, Zimmer SG (2010) Suberoylanilide hydroxamic acid (SAHA) potentiates paclitaxel-induced apoptosis in ovarian cancer cell lines. Gynecol Oncol 116:126–130
Cooper AL, Greenberg VL, Lancaster PS, Van Nagell JR Jr, Zimmer SG, Modesitt SC (2007) In vitro and in vivo histone deacetylase inhibitor therapy with suberoylanilide hydroxamic acid (SAHA) and paclitaxel in ovarian cancer. Gynecol Oncol 104:596–601
Dowdy SC, Jiang S, Zhou XC, Hou X, Jin F, Podratz KC, Jiang SW (2006) Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol Cancer Ther 5:2767–2776
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55
Blagosklonny MV, Robey R, Sackett DL, Du L, Traganos F, Darzynkiewicz Z, Fojo T, Bates SE (2002) Histone deacetylase inhibitors all induce p21 but differentially cause tubulin acetylation, mitotic arrest, and cytotoxicity. Mol Cancer Ther 1:937–941
Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P (2003) HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J 22:1168–1179
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12:1247–1252
Panday VR, Huizing MT, van Warmerdam LJ, Dubbelman RC, Mandjes I, Schellens JH, Huinink WW, Beijnen JH (1998) Pharmacologic study of 3-hour 135 mg M-2 paclitaxel in platinum pretreated patients with advanced ovarian cancer. Pharmacol Res 38:231–236
Huizing MT, Keung AC, Rosing H, van der Kuij V, ten Bokkel Huinink WW, Mandjes IM, Dubbelman AC, Pinedo HM, Beijnen JH (1993) Pharmacokinetics of paclitaxel and metabolites in a randomized comparative study in platinum-pretreated ovarian cancer patients. J Clin Oncol 11:2127–2135
Gianni L, Kearns CM, Giani A, Capri G, Viganó L, Lacatelli A, Bonadonna G, Egorin MJ (1995) Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J Clin Oncol 13:180–190
Ohtsu T, Sasaki Y, Tamura T, Miyata Y, Nakanomyo H, Nishiwaki Y, Saijo N (1995) Clinical pharmacokinetics and pharmacodynamics of paclitaxel: a 3-hour infusion versus a 24-hour infusion. Clin Cancer Res 1:599–606
Steed H, Sawyer MB (2007) Pharmacology, pharmacokinetics and pharmacogenomics of paclitaxel. Pharmacogenomics 8:803–815
Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP (1999) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401:188–193
Munster PN, Troso-Sandoval T, Rosen N, Rifkind R, Marks PA, Richon VM (2001) The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer cells. Cancer Res 61:8492–8497
Bali P, PranpatM SwabyR, Fiskus W, Yamaguchi H, Balasis M, Rocha K, Wang HG, Richon V, Bhalla K (2005) Activity of suberoylanilide hydroxamic acid against human breast cancer cells with amplification of her-2. Clin Cancer Res 11:6382–6389
Zhou Q, Shaw PG, Davidson NE (2009) Inhibition of histone deacetylase suppresses EGF signaling pathways by destabilizing EGFR mRNA in ER-negative human breast cancer cells. Breast Cancer Res Treat 117:443–451
De los Santos M, Martínez-Iglesias O, Aranda A (2007) Anti-estrogenic actions of histone deacetylase inhibitors in MCF-7 breast cancer cells. Endocr Relat Cancer 14:1021–1028
Sonnemann J, Gänge J, Pilz S, Stötzer C, Ohlinger R, Belau A, Lorenz G, Beck JF (2006) Comparative evaluation of the treatment efficacy of suberoylanilide hydroxamic acid (SAHA) and paclitaxel in ovarian cancer cell lines and primary ovarian cancer cells from patients. BMC Cancer 6:183
Ruefli AA, Bernhard D, Tainton KM, Kofler R, Smyth MJ, Johnstone RW (2002) Suberoylanilide hydroxamic acid (SAHA) overcomes multidrug resistance and induces cell death in P-glycoprotein-expressing cells. Int J Cancer 99:292–298
Peart MJ, Tainton KM, Ruefli AA, Dear AE, Sedelies KA, O’Reilly LA, Waterhouse NJ, Trapani JA, Johnstone RW (2003) Novel mechanisms of apoptosis induced by histone deacetylase inhibitors. Cancer Res 63:4460–4471
Marchion D, Münster P (2007) Development of histone deacetylase inhibitors for cancer treatment. Expert Rev Anticancer Ther 7:583–598
Tan J, Cang S, Ma Y, Petrillo RL, Liu D (2010) Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J Hematol Oncol 3:5
Chou TC (2006) Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58:621–681
Torres K, Horwitz SB (1998) Mechanisms of Taxol-induced cell death are concentration dependent. Cancer Res 58:3620–3626
Luong QT, O’Kelly J, Braunstein GD, Hershman JM, Koeffler HP (2006) Antitumor activity of suberoylanilide hydroxamic acid against thyroid cancer cell lines in vitro and in vivo. Clin Cancer Res 12:5570–5577
Blagosklonny MV, Schulte TW, Nguyen P, Mimnaugh EG, Trepel J, Neckers L (1995) Taxol induction of p21WAF1 and p53 requires c-raf-1. Cancer Res 55:4623–4626
Etienne-Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420:629–635
Wittmann T, Waterman-Storer CM (2001) Cell motility: can Rho GTPases and microtubules point the way? J Cell Sci 114:3795–3803
Niculescu AB III, Chen X, Smeets M, Hengst L, Prives C, Reed SI (1998) Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol 18:629–643
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 21072105) and the Independent Innovation Foundation of Shandong University (No.2009DX002).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shi, Yk., Li, Zh., Han, Xq. et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances taxol-induced cell death in breast cancer. Cancer Chemother Pharmacol 66, 1131–1140 (2010). https://doi.org/10.1007/s00280-010-1455-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-010-1455-1