
Abstract
Cancer is a serious and life-eliminating disease. Majority of anticancer agents are non-selective. Along with the cancerous cells they also target the normal ones. An important aspect is to hit the developing mechanism of the tumor, which is highlighted by in silico drug designing. On the basis of novel molecular targets, in silico (computational approach) drug discovery has emerged as today’s need. Histone deacetylases are an important therapeutic target for many human cancers. The first and only approved (in 2006) histone deacetylase inhibitors (HDACIs) is Zolinza. Depending on the types of the histone deacetylase (HDAC) enzymes, discovery of type-specific inhibitors is important. With continued research and development, in near future HDACIs are likely to figure prominently in cancer treatment plans. This review presents the overview of HDACs, their role in cancer, their structural classes, activity, catalytic domain and the inhibitors of HDACs for cancer therapy. Also it helps in understanding the open directions in this area of research and highlights the importance of computational approaches in discovering specific drugs for cancer therapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
From the beginning, human life has been threatened by various diseases. Among them, cancer is a serious and pathogenic one. Great progress has been made for the treatment of cancer in recent years [1]. The majority of anticancer drugs are cytotoxic agents with low therapeutic index acting in a non-selective manner against proliferative cells of both cancerous and normal tissues [2]. The production of more effective anticancer drugs with novel modes of action is today’s need [3, 4]. In order to improve the therapeutic efficiency, oncologists are aware of the fact that new drug discovery must target the developing mechanism of tumor [1]. Many transcription-regulating proteins are themselves deregulated in cancer at the level of expression or activity. Histone deacetylases (HDACs) is one such family of proteins [5].
Along with the mutations in the DNA code cancer can result from aberrations in chromatin-modifying proteins such as HDACs and chromatin [5]. Disruption of histone acetyl transferases (HAT) or HDAC activity may play an important role in the uncontrolled cell growth of cancer [6]. In the discovery of drugs, HDAC has become a novel target for the treatment of cancer and other diseases [7–12]. Over the past few years, the number of HDAC enzyme subtypes has expanded considerably offering opportunities for the development of HDAC inhibitors with improved specificity [13]. With continued research and development, in near future histone deacetylase inhibitors (HDACIs) are likely to figure prominently in cancer treatment plans [5].
The first and only approved (in 2006) HDAC inhibitor Zolinza by US Food and Drug Administration is used for the treatment of cutaneous T cell Lymphoma [5]. In general, HDACIs are associated with certain toxicities [14, 15] and adverse side effects like bone marrow depression, adverse effects to gastrointestinal tract [1], fatigue, vomiting, nausea and diarrhea [5]. In vitro Tubacin selectively targets HDAC6 and it can induce apoptosis of multiple myeloma cells [16]. However, the toxicity profile of it is yet not known and it has yet to enter clinical trials [5].
As HDACs are an important therapeutic target for many human cancers, so it is necessary to design and identify target-specific inhibitors. Depending on the types of the HDAC enzymes, discovery of type-specific inhibitors is important. On the basis of novel molecular targets like HDACs, rational drug discovery has emerged as today’s need [2]. The cytostatic effects that are helpful in reducing normal tissue toxicity rather than cytotoxic effects may be produced by the rational drug discovery of mechanism-based drugs [2].
This review presents the overview of HDACs, their role in cancer, their structural classes, activity, catalytic domain and the inhibitors of HDACs for cancer therapy. Also it helps in understanding the open directions in this area of research and highlights the importance of in silico approaches in discovering specific drugs for cancer therapy.
Histone deacetylases
Histone deacetylases are a family of enzymes involved in DNA repair, regulation of gene expression and stress response. The basic repeating units that constitute the eukaryotic chromatin are nucleosomes. Nucleosome is comprised of an octamer of the four pairs of core histones H2A, H2B, H3 and H4, and ~146 base pairs of DNA wrapped around them [17]. The N-terminal domains of core histones are rich in positively charged basic amino acids, which can actively interact with DNA [18]. Organization of chromatin is important for the regulation of gene expression [19]. HDACs alter the chromatin structure and dynamically affect transcriptional regulation by acetylating and deacetylating the chromatin histone protein [20]. Due to the higher order chromatin structure and the packaging of DNA in nucleosomes, the ability of transcriptional machinery to their target genes is blocked. In order to regulate transcription, the histone tails within the chromatin pass through multiple modifications [20]. One of the major regulatory mechanisms for the gene expression is the balance between the competing activities of HAT and HDACs [21, 22]. The ε-amino groups of conserved lysine residues within the core histones get the acetyl groups from acetyl coenzyme A by means of HATs [23]. By neutralizing the positive charge, acetylation loosens the interaction of histones with negatively charged DNA backbone, thus favoring the binding of transcription factors (TFs) for active gene transcription and leading to a more open active chromatin structure [24]. The tightened interaction between the DNA and histones, which inhibits gene transcription by blocking the binding sites on promoter is thought to be due to the re-establishment of the positive charge in the amino-terminal tails of core histones catalyzed by HDACs [25]. Transcriptional repression is associated with low levels of histone acetylation whereas transcriptionally active genes are associated with highly acetylated core histones [26, 27]. The activities of two enzyme families, the HATs and HDACs control the acetylation status of histones [28]. Internucleosomal histone–histone interactions and association of other proteins with chromatin are affected by deacetylation. By changing gene expression patterns in the absence of mutations of genome itself, HDACs influence the epigenetic status of the cell [5]. It has been discovered that many cancers occur by a genome-wide histone hypoacetylation [29].
In order to avoid the dramatic effects such as carcinogenesis on cell phenotype, the normal balance between the actions of HATs and HDACs is necessary [14].
Structural classes of HDACs
Structurally and functionally 18 different known human HDACs have been grouped into two families and four classes [30]. The two families are Classical and Sirtuin family.
Class I, class IIa, class IIb and class IV are included in the classical family whereas the Sirtuin family contains class III HDACs [5]. Class I comprises of HDAC1, HDAC2, HDAC3 and HDAC8 whereas class IIa has HDAC4, HDAC5, HDAC7 and HDAC9. Class IIb contains HDAC6 and HDAC10. Class IV has only HDAC11 [5].
Class I and class II HDACs are different from class III in respect of cofactor requirements [31]. Class I and II have zinc as a cofactor so they are hydrolases which contain zinc. Class III which are a series of NAD-dependent Sir2 family differs from other HDAC classes [32]. Class I and II HDACs are mainly found to be involved in cancer pathogenesis [19]. Class I are found exclusively in nucleus whereas on certain cellular signals class II shuttle between the nucleus and cytoplasm [19]. TFs including p53 and non-histone proteins are deacetylated by Class III HDACs [31, 33].
In regulating cell proliferation, Class I HDACs are more significant than the Class II enzymes [34]. Suppression of apoptosis in cancer cells might be due to HDAC2 [35]. In cardiomyocyte differentiation HDAC5 and HDAC9 play a role [36]. Majority of HDACIs are unlikely to be isoform-specific [37].
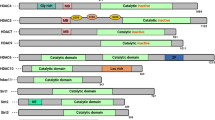
HDAC8 is, to date, the only mammalian isoform for which a crystal structure is available as shown in Fig. 1 [38] and due to the lack of availability of the crystal structures rational design is being hindered [39].

Crystal structure of HDAC8
HDACs catalytic domain
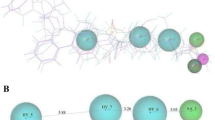
The catalytic domain of HDACs consists of a narrow tube-like pocket having the length equivalent to 4–6 carbon straight chains. Near the bottom of the enzyme pocket a Zn+2 ion is positioned [40], which facilitates the deacetylation catalysis in cooperation with two His-Asp charge relay systems [31]. Unusually, HDAC6 and HDAC10 have two catalytic domains among the known human HDAC enzymes and both domains of HDAC6 are used for deacetylase activity [41]. The binding of suberoylanilide hydroxamic acid (SAHA) has been shown to the catalytic domain of the HDAC enzyme has been shown in Fig. 2 [7].

SAHA binds to the cavity of HDAC enzyme. The hydroxamic acid moiety of SAHA binds to zinc atom (pink), allowing the rest of the molecule to lie along the surface of the enzyme
HDACs malfunctioning disorders
Class I HDACs are mostly confined to the nucleus whereas classes II HDACs are transported between nucleus and cytoplasm [42]. The roles of class 1 HDACs in cell survival and proliferation whereas those of class II HDACs in having tissue-specific roles have been obtained through knock out analysis [43]. Through direct interaction with the TF HDAC1 and HDAC3 enhance hypoxia-inducible factor-1a stability [44]. By regulation of p53-binding activity HDAC2 modulates transcriptional activity [45]. Cardiac defects are caused due to HDAC2 knockouts [46]. Also HDAC5 and HDAC9 knockouts results in cardiac defects [36]. HDAC7 knockouts cause defects in the maintenance of vascular integrity [47]. The detailed mechanism of how the malfunctioning in HDACs results in various disorders is not clear, because little is known about the sequence specificity of histones that determine the activity of different HDACs [48].
HDACs were ranked according to their substrate selectivity using a library of fluorigenic tetrapeptide substrates as HDAC8 > HDAC1 > HDAC3 > HDAC6 [49]. In cancer, structural mutations in HDACs are rare. However, in various cancers, changes in expression of different HDACs have been reported. In colon cancer samples, HDAC2 and HDAC3 proteins are increased [42]. In gastric cancer, HDAC1 is increased. In lung cancer, reduced expression of HDAC5 and HDAC10 is associated with poor scenario.
HDACs and HATs interact with DNA through multiprotein complexes that include co-repressors and co-activators and hence do not bind to it directly [50].
HDAC inhibitors
Histone deacetylases inhibitors have recently emerged as an important class of anticancer agents [51]. HDACIs along with their antitumor effects may also potentiate cytotoxic agents or synergize with other targeted anticancer agents [52].
HDAC inhibitors target the gene expression without modifying the DNA sequence. The transcription of several tumor suppressor genes is prevented when HDACs bind DNA tightly to histones [53]. HDAC inhibitors are potent inducers of differentiation, growth arrest, and apoptosis of transformed cells through regulation of gene expression [54]. In cell cycle, apoptosis and DNA synthesis, a common set of genes regulated by all HDAC inhibitors was found to be predominantly involved [55]. The substrate access is blocked, causing an accumulation of acetylated histones when HDAC inhibitors bind directly to the HDAC-active site [56].
Most of anticancer drugs when were used to treat tumor cells have shown side effects like myocardium damage and bone marrow depression even leading to cell death [57].
Three-component concept
The wide variety of structural HDAC inhibitors include molecular fragments, a linker domain which occupies the channel, a metal-binding domain that interacts with the active site and surface recognition domain that interacts with the residues on the rim of the active site. In developing potent HDAC inhibitors, this three-component concept has proved to be successful [58].
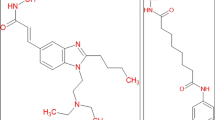
This three-component concept has also been defined as that the inhibitors possess a cap group, an aliphatic chain and a functional group that would chelate the metal cation in the active site as explained by an example of Trichostatin A (TSA) in Fig. 3 [59]. TSA is an HDAC inhibitor [57]. This pharmacophore model may provide guidance for the rational design to discover novel HDAC inhibitors by highlighting the important binding features of HDAC ligands [20].

Properties of HDAC inhibitors
Structural classes of HDACIs
The HDACIs comprises of four main structural classes and represent a broad family of chemical compounds [5]:
-
Hydroxamates
-
Benzamides
-
Short-chain fatty acids
-
Cyclic peptides
Hydroxamates
The largest class of HDAC inhibitors with great therapeutic potential is hydroxamic acids [19].
The first discovered natural product that belongs to hydroxamates group is Trichostatin A (TSA) [60]. TSA is a frequent HDAC inhibitor [57]. TSA was originally isolated from Streptomyces hygroscopicus as antifungal antibiotic active against Trichophyton [61]. The structural analog, SAHA of TSA and its low concentration, (nM) inhibit growth in tumors and can induce cell differentiation with little effects on normal cells [62].
Recent studies have shown that SAHA and TSA inhibit the activity of Class I and II HDACs and are able to induce cell differentiation of lymphoma [63]. TSA is less efficient against HDAC8 and inhibits HDAC1, -2, -3 and HDAC4, -6, -7 and -9 HDACs [64]. Class III HDACs (sirtuins) that are NAD+-dependent are not affected by TSA treatment. The chemical structure of TSA is characterized by a 4-(N, N-dimethylamino) phenyl moiety linked through a carbonyl group to an aliphatic unsaturated chain (4, 6-dimethyl-2, 4-hexanedienoic acid) bearing at the end a hydroxamate portion. At C6 position, TSA possesses a chiral center [65].
In TSA, biochemical mechanism of enzyme inhibition, the 4-(N,N-dimethylamino)phenyl moiety of TSA works as a cap to pack the inhibitor at the rim of the tubular active site pocket of the enzyme, and the hydroxamic acid function chelates the zinc ion in a bidentate fashion at the bottom of the pocket determining the inhibitory effect [66].
According to the latest studies, telomerase activity is also inhibited by TSA. Although TSA has a wide range of anticancer effects, but due to its side effects, it has not so far been used in clinical trials [67].
Chemically SAHA and related HDACIs contain (1) a hydrophobic CAP group, able to interact with the rim of the catalytic tunnel of the enzyme, (2) a polar connection unit (CU), present not in all but in most HDACIs, which can make hydrogen bonds with some amino acid residues in the tunnel, (3) a 4- to 6-carbon unit hydrophobic spacer (linker), which allows to the following (4) zinc-binding group (i.e., the hydroxamate group in SAHA and TSA) to reach and to complex the zinc ion, and thus to inhibit the enzyme [56, 68, 69].
SAHA displays lower potency against HDAC8 and efficiently inhibits HDAC1, -2, -3, -4, -6, -7 and -9 analogously to TSA [64]. The mechanisms underlying its anticancer activity are still not fully characterized and are very complex.
Vorinostat involve the accumulation of acetylated histones resulting in transcriptional activation in part with the antiproliferative effects [70]. In its anticancer activity, vorinostat also uses non-histone proteins including TFs as substrates for HDACs, and the effects of it on these proteins might be also important [70].
Panobinostat (LBH-589) is less potent against HDAC6 and one or two magnitudes less active against HDAC8, but is highly active against HDAC1, -2, -3, -4, -7, -9 [65].
Belinostat that inhibited both class I and class II HDACs in the submicromolar range is another cinnamyl hydroxamate. For hematological malignancies as well as for many solid tumors belinostat is in Phase II clinical trials [71].
Benzamides
Having MS-275 being the lead compound, a series of benzamide derivatives with a chemical structure unrelated to the other synthetic HDACIs was described [72]. As suggested by molecular modeling studies, these compounds showed a 2-aminoanilide moiety at the right end of the molecule, which likely can function as a weak zinc-chelating group into the tube-like active site of the deacetylase core of the enzyme [73], or may contact key amino acids in the active site without Zn-ion coordination.
MS-275 is inactive against HDAC4, -6, -7 and -8 and inhibits preferentially HDAC1 and -9, HDAC2 and -3 [64]. Orally administered MS-275 strongly inhibited the growth in seven of eight tumor lines implanted into nude mice, most of which not responding to 5-fluorouracil [74].
The inhibitors of class III HDACs (sirtuins) in contrast to class I/II/IV HDACs are much less validated as anticancer agents and in general less developed on the pharmacological side. In many cellular processes, such as regulation of TFs, gene silencing, lifespan extension, cell-cycle regulation and fatty acid metabolism, sirtuins play an important role [32].
MS-275 and CI-994 that are benzamide HDAC inhibitors are now in Phase I and II clinical trials. MS-275 has little effect on HDAC-8 while it preferentially inhibits HDAC1 over HDAC3 [75]. The synergistic effects with other therapeutic strategies and the practical uses of HDAC inhibitors in cancer therapy are yet unresolved [76].
Short-chain fatty acids
Sodium butyrate, belonging to the natural short-chain fatty acid, which is fermental production of bacterium in colon, is the first confirmed HDAC inhibitor. Butyrate, valproic acid, phenylbutyrate and their derivatives belong to aliphatic acid group. In clinical application, this group has been limited due to its high effective millimolar concentrations when assessed in cells [75].
The short-chain fatty acids mechanism of action as HDACIs has yet not been fully elucidated [65]. Because of their short side chains, short-chain fatty acids have low potency, which limits their contact with the catalytic pocket of HDACs [77]. In human prostate cancer and cervical carcinoma cells, butyrates inhibit the growth, while in acute myeloid leukemia, butyrates induce granulocyte maturation and histone acetylation [78].
In both hematological malignancies and solid tumor lines, butyrates have been under clinical evaluation [79]. Butanoic acid is currently under clinical trials as in leukemia tumor cell lines, it showed tenfold more potency than sodium butyrate [80].
Although the activity of short-chain fatty acids is weak but among them various have entered the clinical trials because of their use for alternative medical conditions [81, 82].
Cyclic peptides
A number of natural products with significant in vitro activity as trapoxin A and B and depudecin are the HDAC inhibitors which belong to this class [79]. Trapoxin is a hybrid of cyclic peptide and epoxyketone moiety [83].
Depsipeptide, a natural product extracted from Chromobacterium violaceum is another HDAC inhibitor, which belongs to the cyclic peptides group [84]. For cancer therapy, it has been under the multiple Phase I and II trials. It inhibits HDACs at nanomolar concentrations [79].
Important compounds of HDAC inhibitors and their structures [85], which are under clinical trials, are summarized in Table 1.
In silico approach and type-specific HDAC inhibitor identification
Use of computational techniques is getting rapidly involved in the drug discovery and development process [86]. Various terms that include in silico drug design, rational drug design, structure-based drug design, computer-aided molecular modeling and computational drug design are being used for this area [86]. In silico or rational drug designing is a systematic process, which involves a number of predefined steps [87]. Identification of the relevant target molecule is defined to be the first step in this process [88]. Developments in bioinformatics are helpful in identifying excellent drug targets [89]. The target that is closely linked to human disease is considered to be an ideal one for in silico drug designing [89]. Mostly the good targets for the drugs are proteins [89]. In some case, enzymes are also the excellent drug targets because compounds that can fit in the active site can be designed [89].
Recent research showed that in drug discovery, HDAC has become a novel target for the treatment of cancer [1]. The number of HDAC enzyme subtypes has expanded considerably offering opportunities for the development of HDAC inhibitors with improved specificity [13]. Crystal structure of HDAC8 is the only mammalian isoform available to date [38]. So there is a need to identify the crystal structure of all possible isozymes in order to identify the specific HDAC inhibitors. HDACs are an important therapeutic target for cancer so they can be used for in silico drug designing.
After target identification, the second step of in silico approach is the selection of the most suitable molecule (Lead) that binds tightly to the target, has no side effects like toxicity, is bio-accessible and can be synthesized easily [90]. Lead identification is the most critical stage in drug discovery [2]. The discovery and development of isozyme-specific HDAC inhibitors or Lead would be of both research and clinical interests. With continued research and development, in near future HDACIs are likely to figure prominently in cancer treatment plans [5]. Most of the HDAC inhibitors that are known or are in clinical trials are unselective, or are just partially selective for one class of HDACs [14, 15]. Consequently, there is a need to identify selective Lead for the specific isoforms of HDACs. More focused cancer therapies and new clinical opportunities would also be provided by selective HDAC inhibitors. It is not clear whether the antitumor properties of HDAC inhibitors are the consequence of targeting one or few “crucial” subtypes or are due to their lack of specificity. This question is of considerable interest because it opens the way for the development of more selective novel compounds, possibly with enhanced efficacy and tolerability [91].
The first and only approved HDAC inhibitor by US Food and drug administration is SAHA [5]. SAHA is approved under the trade name Zolinza, for treatment of cutaneous T cell Lymphoma [5]. Only one specific HDAC inhibitor is currently available [5]. Tubacin has the ability of selectively targeting HDAC6; induces apoptosis in myeloma cells and also inhibit transforming growth factor β-induced epithelial-to-mesenchymal transition [5, 16]. Identification of type-specific HDAC inhibitors is necessary and will reduce toxic side effects.
The greater opportunity for in silico drug design, in discovering new leads is being fueled by the structural genomics revolution, start of proteomics, completion of human genome project and the developments in information technology [89]. In silico approach of drug designing in identifying the promising candidates has proved to be the faster one with the advantage of cost minimization and discarding the non-starters before too much of the money has been wasted [2]. Early anticancer agents were discovered largely serendipitously [92]. The anticipation about majority of future anticancer drugs is that they will be significantly less toxic and target-specific [93]. By applying in silico drug discovery techniques, it is anticipated that more potent and specific anticancer agents will be discovered with in the next decade [93].
Conclusion and future directions
A growing interest in the discovery and development of HDAC inhibitors has been observed during the past few years. HDAC enzyme inhibition would be a promising approach for the treatment of human cancers [19].
To date, the one and only approved HDAC inhibitor is Zolinza [5]. So there is a need for more specific HDAC inhibitors for cancer treatment. There has been a constant drive towards the highly target-specific drugs with the introduction of in silico approaches in the domain of drug discovery. More specific compounds would certainly provide interesting pharmacological tools for elucidating functions of individual HDACs. Too many HDAC inhibitors are available and many of them can be screened using assays [19]. So instead of designing newer and newer compounds, there is a need to identify more specific HDAC inhibitors for the specific enzyme types. By using in silico approaches, not only the specific enzyme types can be identified using comparative modeling studies, but also the type-specific inhibitors can be designed. So in lesser time and cost without wasting the important chemicals, the important results can be first identified through computational methods and then after getting the proper lead, it could be synthesized and tested for clinical trials.
We conclude by the remarks that in silico approach has appeared as a road, which can lead us to success in discovering specific HDACIs for the treatment of many human cancers caused by HDACs abnormal functions.
References
Li Q, Xu W (2005) Novel anticancer targets and drug discovery in post genomic age. Curr Med Chem Anticancer Agents 5:53–63
Seddon BM, Workman P (2003) The role of functional and molecular imaging in cancer drug discovery and development. Br J Radiol 76:S128–S138
Sikora K, Advani S, Koroltchouk V, Magrath I, Levy L, Pinedo H, Schwartsmann G, Tattersall M, Yan S (1999) Essential drugs for cancer therapy: a World Health Organization consultation. Ann Oncol 10:385–390
Gelmon KA, Eisenhauer EA, Harris AL, Ratain MJ, Workman P (1999) Anticancer agents targeting signalling molecules and cancer cell environment: challenges for drug development. J Natl Cancer Inst 91:1281–1287
Walkinshaw DR, Yang XJ (2008) Histone deacetylase inhibitors as novel anticancer therapeutics. Curr Oncol 15:237–243
Chen JS, Faller DV, Spanjaard RA (2003) Short-chain fatty acid inhibitors of histone deacetylases: promising anticancer therapeutics. Curr Cancer Drug Targets 3:219–236
Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK (2001) Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 1:194–202
Kelly WK, Connor OAO, Marks PA (2002) Histone deacetylase inhibitors: from target to clinical trials. Expert Opin Investig Drugs 11:1695–1713
Marks PA, Richon VM, Breslow R, Rifkind RA (2001) Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol 13:477–483
Marks PA, Richon VM, Breslow R, Rifkind RA (2001) Inhibitors of histone deacetylase are potentially effective anticancer agents. Clin Cancer Res 7:759–760
Phiel CJ, Zhang F, Huang EY, Guenther MJ, Lazar MA, Klein PS (2001) Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 76:36734–36741
Meinke PT, Liberator P (2001) Histone deacetylase: a target for antiproliferative and antiprotozoal agents. Curr Med Chem 8:211–235
Juvale DC, Kulkarni VV, Deokar HS, Wagh NK, Padhye SB, Kulkarni VM (2006) 3D-QSAR of histone deacetylase inhibitors: hydroxamate analogues. Org Biomol Chem 4:2858–2868
Minucci S, Pelicci PG (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 6:38–51
Karagiannis TC, El-Osta A (2007) Will broad-spectrum histone deacetylase inhibitors be superseded by more specific compounds. Leukemia 21:61–65
Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, Anderson KC (2005) Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci USA 102:8567–8572
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389:251–260
Lenfant F, Mann RK, Thomsen B, Ling X, Grunstein M (1996) All four core histone N-termini contain sequences required for the repression of basal transcription in yeast. EMBO J 15:3974–3985
Kouraklis G, Misiakos EP, Theocharis S (2006) Histone deacetylase inhibitors as a potential therapeutic agent for human cancer treatment. Target Oncol 1:34–41
Chen Y, Jiang YJ, Zhou JW, Yu QS, You QD (2008) Identification of ligand features essential for HDACs inhibitors by pharmacophore modeling. J Mol Graph Model 26:1160–1168
Cress WD, Seto E (2000) Histone deacetylases, transcriptional control, and cancer. J Cell Physiol 184:1e16
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403:41e45
Tanner KG, Trievel RC, Kuo MH, Howard RM, Berger SL, Allis CD, Marmorstein R, Denu JM (1999) Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J Biol Chem 274:18157–18160
Gregory PD, Wagner K, Horz W (2001) Histone acetylation and chromatin remodeling. Exp Cell Res 265:195–202
Roth SY, Denu JM, Allis CD (2001) Histone acetyltransferases. Annu Rev Biochem 70:81–120
Pazin MJ, Kadonaga JT (1997) What’s up and down with histone deacetylation and transcription. Cell 89(3):325–328
Grunstein M (1997) Histone acetylation in chromatin structure and transcription. Nature 389:349–352
Struhl K (1998) Histone acetylation and transcriptional regulatory mechanisms. Genes Dev 12:599–606
Mahlknecht U, Hoelzer D (2000) Histone acetylation modifiers in the pathogenesis of malignant disease. Mol Med 6:623–644
Khochbin S, Verdel A, Lemercier C, Seigneurin-Berny D (2001) Functional significance of histone deacetylase diversity. Curr Opin Genet Dev 11:162–166
Lin HY, Chen CH, Lin SP, Weng JR, Chen CH (2006) Targeting histone deacetylase in cancer therapy. Med Res Rev 26(4):397–413
Blander G, Guarente L (2004) The Sir2 family of protein deacetylases. Annu Rev Biochem 73:417–435
Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W (2001) Negative control of p53 by Sir2 alpha promotes cell survival under stress. Cell 107(2):137–148
Park JH, Jung Y, Kim TY, Kim SG, Jong HS, Lee JW, Kim DK, Lee JS, Kim NK, Kim TY et al (2004) Class I histone deacetylase selective novel synthetic inhibitors potently inhibit human tumor proliferation. Clin Cancer Res 10:5271–5281
Zhu P, Huber E, Kiefer F, Gottlicher M (2004) Specific and redundant functions of histone deacetylases in regulation of cell cycle and apoptosis. Cell Cycle 3:1240–1242
Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN (2002) Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110:479–488
Inche A, La Thangue NB (2006) Chromatin control and cancer drug discovery: realising the promise. Drug Discov Today 11:97–109
Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW (2004) Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases structure. Structure 12:1325–1334
Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkuhler C, Marco SD (2004) Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci USA 101:15064–15069
Chen Y, Li H, Tang W, Zhu C, Jiang Y, Zou J, Yu Q, You Q (2009) 3D-QSAR studies of HDACs inhibitors using pharmacophore-based alignment. Eur J Med Chem 44(7):2868–2876
Zhang Y, Gilquin B, Khochbin S, Matthias P (2006) Two catalytic domains are required for protein deacetylation. J Biol Chem 281:2401–2404
Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5:769–784
Lagger G, O’Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, Seiser C (2002) Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J 21:2672–2681
Kim SH, Jeong JW, Park JA, Lee JW, Seo JH, Jung BK, Bae MK, Kim KW (2007) Regulation of the HIF-1a stability by histone deacetylases. Oncol Rep 17:647–651
Harms KL, Chen X (2007) Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res 67:3145–3152
Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, Ferrari VA, Abrams CS, Gruber PJ, Epstein JA (2007) Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3h activity. Nat Med 13:324–331
Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN (2006) Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 126:321–334
Dokmanovic M, Clarke C, Marks PA (2007) Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res 5(10):981–989
Riester D, Hildmann C, Grune wald S, Beckers T, Schwienhorst A (2007) Factors affecting the substrate specificity of histone deacetylases. Biochem Biophys Res Commun 357:439–445
Marmorstein R (2001) Structure of histone acetyltransferases. J Mol Biol 311:433–444
Elaut G, Rogiers V, Vanhaecke T (2007) The pharmaceutical potential of histone deacetylase inhibitors. Curr Pharm Des 13:2584–2620
Sigalotti L, Fratta E, Coral S, Cortini E, Covre A, Nicolay HJ, Anzalone L, Pezzani L, Di Giacomo AM, Fonsatti E, Coalizzi F, Altomonte M, Calabro L, Maio M (2007) Epigenetic drugs as pleiotropic agents in cancer treatment: biomolecular aspects and clinical applications. J Cell Physiol 212:330–344
Garber K (2004) Purchase of Aton spotlights HDAC inhibitors. Nat Biotechnol 22:364–365
Fang JY (2005) Histone deacetylase inhibitors, anti-cancerous mechanism and therapy for gastrointestinal cancers. J Gastroenterol Hepatol 20:988–994
Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG, Davidsen SK (2003) Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol Cancer Ther 2:151–163
Miller TA, Witter DJ, Belvedere S (2003) Histone deacetylase inhibitors. J Med Chem 46:5097–5116
Bi G, Jiang G (2006) The molecular mechanism of HDAC Inhibitors in anticancer effects. Cell Mol Immunol 3(4):285–290
Jung M, Brosch G, Kölle D, Scherf H, Gerhäuser C, Loid P (1999) Amide analogues of trichostatin A as inhibitors of histone deacetylase and inducers of terminal cell differentiation. J Med Chem 42:4669–4679
Grozinger CM, Schreiber SL (2002) Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem Biol 9:3–16
Yoshida M, Kijima M, Akita M, Beppu T (1990) Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem 265:17174–17179
Tsuji N, Kobayashi M, Nagashima K, Wakisaka Y, Koizumi K (1976) A new antifungal antibiotic, trichostatin. J Antibiot (Tokyo) 29:1–6
Marks PA, Dokmanovic M (2005) Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs 14:1497–1511
Wegener D, Hildmann C, Schwienhorst A (2003) Recent progress in the development of assays suited for histone deacetylase inhibitor screening. Mol Genet Metab 80:138–147
Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M (2007) Determination of the class and isoform selectivity of small molecule HDAC inhibitors. Biochem J 409:581–589. doi:10.1042/BJ20070779
Maia A, Altucci L (2009) Epi-drugs to fight cancer: from chemistry to cancer treatment, the road ahead. Int J Biochem Cell Biol 41:199–213
Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP (1999) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401:188–193
Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, Marks PA (1998) A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci USA 95:3003–3007
Mai A, Massa S, Pezzi R, Simeoni S, Rotili D, Nebbioso A, Scognamiglio A, Altucci L, Loidl P, Brosch G (2005) Class II (IIa)-selective histone deacetylase inhibitors. 1. Synthesis and biological evaluation of novel (aryloxopropenyl)pyrrolyl hydroxyamides. J Med Chem 48:3344–3353
Paris M, Porcelloni M, Binaschi M, Fattori D (2008) Histone deacetylase inhibitors: from bench to clinic. J Med Chem 51:1505–1529
Kelly WK, Marks PA (2005) Drug insight: histone deacetylase inhibitors—development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat Clin Pract Oncol 2:150–157
Qian X, Ara G, Mills E, LaRochelle WJ, Lichenstein HS, Jeffers M (2008) Activity of the histone deacetylase inhibitor belinostat (PXD101) in preclinical models of prostate cancer. Int J Cancer 122:1400–1410
Suzuki T, Ando T, Tsuchiya K, Fukazawa N, Saito A, Mariko Y, Yamashita T, Nakanishi O (1999) Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J Med Chem 42:3001–3003
Wang DF, Helquist P, Wiech NL, Wiest O (2005) Toward selective histone deacetylase inhibitor design: homology modeling, docking studies, and molecular dynamics simulations of human class I histone deacetylases. J Med Chem 48:6936–6947
Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O (1999) A synthetic inhibitor of histone deacetylase, MS-27–275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci USA 96:4592–4597
Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, Macarron R, Los Frailes MD, Perez P, Krawiec J, Winkler J, Jaye M (2003) Identification of novel isoformselective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther 307:720–728
Pan L, Lu J, Huang B (2007) HDAC inhibitors: a potential new category of anti-tumor agents. Cell Mol Immunol 4(5):337–343
Johnstone RW (2002) Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov 1:287–299
Finzer P, Stohr M, Seibert N, Rosl F (2003) Phenylbutyrate inhibits growth of cervical carcinoma cells independent of HPV type and copy number. J Cancer Res Clin Oncol 129:107–113
Acharya MR, Sparreboom A, Venitz J, Figg WD (2005) Rational development of histone deacetylase inhibitors as anti-cancer agents: a review. Mol Pharmacol 68:917–932
Batova A, Shao LE, Diccianni MB, Yu AL, Tanaka T, Rephaeli A, Nudelman A, Yu J (2002) The histone deacetylase inhibitor AN-9 has selective toxicity to acute leukemia and drug-resistant primary leukemia and cancer cell lines. Blood 100:3319–3324
Gore SD, Weng LJ, Figg WD, Zhai S, Donehower RC, Dover G, Grever MR, Griffin C, Grochow LB, Hawkins A, Burks K, Zabelena Y, Miller CB (2002) Impact of prolonged infusions of the putative differentiating agent sodium phenylbutyrate on myelodysplastic syndromes and acute myeloid leukemia. Clin Cancer Res 8:963–970
Gore SD, Weng LJ, Zhai S, Figg WD, Donehower RC, Dover GJ, Grever M, Griffin CA, Grochow LB, Rowinsky EK, Zabalena Y, Hawkins AL, Burks K, Miller CB (2001) Impact of the putative differentiating agent sodium phenylbutyrate on myelodysplastic syndromes and acute myeloid leukemia. Clin Cancer Res 7:2330–2339
Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T (1993) Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J Biol Chem 268:22429–22435
Nakajima H, Kim YB, Terano H, Yoshida M, Horinouchi S (1998) FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp Cell Res 241:126–133
Andrianov V, Gailite V, Lola D, Loza E, Semenikhina V, Kalvinsh I, Finn P, Petersen KD, Ritchie JWA, Khan N, Tumber A, Collins LS, Vadlamudi SM, Börkling F, Sehested M (2009) Novel amide derivatives as inhibitors of histone deacetylase: design, synthesis and SAR. Euro J Med Chem 44:1067–1085
Kapetanovic IM (2008) Computer-aided drug discovery and development (CADDD): in silico-chemico-biological approach. Chem Biol Interact 171(2):165–176
Garrett MD, Workman P (1999) Discovering novel chemotherapeutic drugs for the third millennium. Eur J Cancer 35:2010–2030
Workman P (2001) New drug targets for genomic cancer therapy: successes, limitations, opportunities and future challenges. Curr Cancer Drug Targets 1:33–47
Anderson AC (2003) The process of structure-based drug design. Chem Biol 10:787–797
Lengauer T, Zimmer R (2000) Protein structure prediction methods for drug design. Brief Bioinform 1(3):275–288
Dallavalle S, Cincinelli R, Nannei R, Merlini L, Morini G, Penco S, Pisano C, Vesci L, Barbarino M, Zuco V, Cesare MD, Zunino F (2009) Design, synthesis, and evaluation of biphenyl-4-yl-acrylohydroxamic acid derivatives as histone deacetylase (HDAC) inhibitors. Eur J Med Chem. doi:10.1016/j.ejmech.2008.11.005
Pratt WB, Ruddon RW, Ensminger WD, Maybaum J (1994) Some milestones in the development of cancer chemotherapy. In: The anticancer drugs, 2nd edn. Oxford University Press, Oxford, pp 17–25
Liu R, Hsieh CY, Lam KS (2004) New approaches in identifying drugs to inactivate oncogene products. Semin Cancer Biol 14:13–21
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Noureen, N., Rashid, H. & Kalsoom, S. Identification of type-specific anticancer histone deacetylase inhibitors: road to success. Cancer Chemother Pharmacol 66, 625–633 (2010). https://doi.org/10.1007/s00280-010-1324-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-010-1324-y